 2018, Vol. 29
2018, Vol. 29 



Purine nucleoside derivatives play important roles in the field of chemical, biological and medicinal research. Some purine nucleoside analogues have been developed as anti-HIV and anti-cancer drugs [1]. Among them, N-9 substituted purine nucleoside analogues are a range of significant heterocyclic compounds involved in biological events [2]. These compounds serve as basic units of nucleic acids and coenzymes, and they are also key constituents in life processes such as metabolism, energy storage and cell signalling [3]. Moreover, there are a number of aminated tetrahydrofuran (THF) derivatives used as therapeutical agents (Fig. 1) [4].

|
Download:
|
| Fig. 1. Selected examples of aminated THF derivatives that exhibit biological activities. | |
To gain better understanding of these purine nucleoside derivatives, pure and structurally well-defined materials are highly desirable. Alkylation is still a major approach to prepare purine nucleoside derivatives, and the most widely-used donors include halogenated compounds [5], mesylates [6], and tosylates [7]. However, only a handful of studies have been done on the synthesis of purine nucleoside analogues via reaction of the sp3 N-H bond of the purine ring and the sp3 C-H of alkyl ether. Guo and co-workers developed the metal-free direct alkylation of purines with THF and ethers, but the reaction required high-power-light illumination and high temperature [8]. Hu and co-workers reported an elegant method for the metal-free direct C-H amination of ethers using a hypervalent iodine reagent at room temperature [9]. Liu and co-workers reported the peroxide-promoted coupling of nucleobases with ethers employing copper-catalyzed pathway, whereas requirements of metal, ligand and high excess of peroxide made this method complicated [10]. Besides, other groups also reported interesting work based on reaction of the sp3 N-H bond and the sp3 C-H of alkyl ether [11]. However, purines were excluded from the substrates, probably due to their low reactivity when compared with other substrates. Meanwhile, regiospecific alkylation (N-9 or N-7) of purine is another challenge, thus the site-selective alkylation of purine analogues at the N-9 position is in great demand.
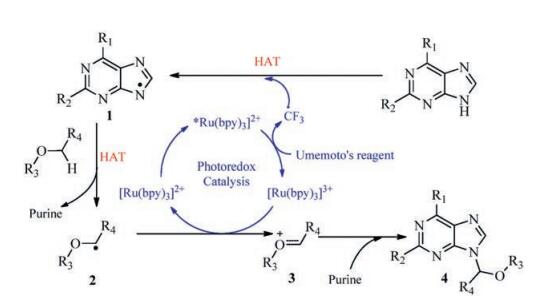
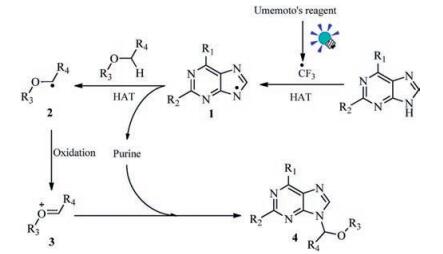
To tackle these problems, we therefore hypothesized that an electrophilic CF3 radical-triggered radical relay would probably lead to an alternative approach to the preparation of N-9 alkylated purine nucleoside derivatives. As we know, in recent years, the use of organic photoredox catalysts in a myriad of synthetic transformations has shown a remarkable "renaissance" [12]. We imagined that visible-light-promoted excitation of [Ru(bpy)3]2+ would precede a SET process to Umemoto's reagent to generate an electrophilic CF3 radical [13] and [Ru(bpy)3]3+(Scheme 1). On account of the polar effects [14], the N-radical 1 was generated through a hydrogen atom transfer (HAT) because of CF3 radical, which was sequentially followed by another HAT process to form C-radical 2. The C-radical 2 could be quickly oxidized by [Ru(bpy)3]3+ to an oxocarbenium ion 3[15], which was then trapped by the nucleophilic purine N-9 position, thus forming the N-9 alkylated purine nucleoside derivatives 4.

|
Download:
|
| Scheme 1. Original hypothesis for the synthesis of N-9 alkylated purine nucleoside derivatives. | |
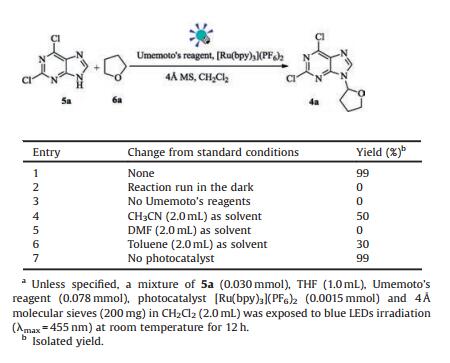
Herein, we report our results inspired by the hypothesis in Scheme 1. Our initial reaction conditions involved irradiation with blue LEDs (λmax = 455 nm) in the presence of 2, 6-dichloropurine 5a (0.030 mmol), tetrahydrofuran (THF) (1.0 mL), Umemoto's reagent (0.078 mmol), photocatalyst [Ru(bpy)3](PF6)2 (0.0015 mmol) and 4 Å molecular sieves in CH2Cl2 (2.0 mL) (Table 1). When the standard conditions were used, the desired product 4a was obtained in nearly quantitative yield. The control experiments demonstrated that the irradiation and the use of Umemoto's reagent are essential. The solvent screening experiments showed the insignificant differences or inferior results from those obtained using CH2Cl2. However, the irradiation without [Ru(bpy)3](PF6)2 provided the similar yield of 99%! This surprising outcome is likely due to the formation of an electron donor-acceptor (EDA) complex [16], in which purine and Umemoto's reagent may serve as the electron donor and electron acceptor, respectively. Thanks to the large conjugated system, the EDA complex could be employed directly as a photon-absorbing EDA that would avoid using photocatalysts.
|
|
Table 1 Initial screening and optimization.a |
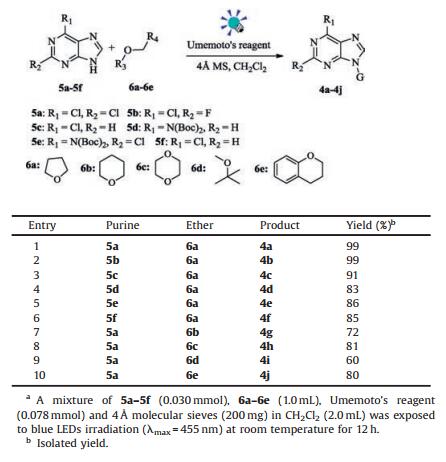
We next investigated the substrate scope of the reaction under the reaction conditions depicted in entry 7 of Table 1. Under the optimized high-yielding reaction conditions, various purines and several other alkyl ethers were examined. As shown in Table 2, various purines were employed as substrates, and different 6-halopurine derivatives 5a–5c could react with THF smoothly (entries 1–3). The purine derivatives with nitrogen-containing groups at the C-6 position (5d–5f) could also afford the alkylated products in good yields (entries 4-6). Besides, it seemed that when the purine derivatives with C-2 position replaced by halogen atoms (such as Cl and F) were used, the yields of products were increased slightly (entries 1, 2 and 5).
|
|
Table 2 Scope of visible-light-promoted synthesis of N-9 alkylated purine nucleoside derivatives.a |
{kind=link}
{kind=link}
After surveying various nucleobases, we turned to test the reactions of various ethers with 2, 6-di-chloropurine 5a under the optimized reaction conditions (Table 2, entries 7–10). To our delight, various ethers were proven to be reliable reactants as illustrated in Table 2. Cyclic ether compounds, either five-(6a) or six-membered ring (6b or 6c), could afford the desired products in good yields (entries 1, 7 and 8). The sterically hindered compound methyl tert-butyl ether (MTBE) (6d) also underwent this reaction to yield the product 4i (entry 9). Interestingly, when the unsymmetrical ether isochroman (6e) was used as substrate, a regioselective product 4j was obtained in good yield (entry 10). The high efficiency of this light-promoted synthesis of N-9 alkylated purine nucleoside derivatives was further highlighted by the irradiation of sunlight. The reaction of 5a and 6a could also give product 4a in good yield (71%, Scheme 2).

|
Download:
|
| Scheme 2. Sunlight-promoted synthesis of the N-9 alkylated purine nucleoside derivative. | |
{kind=link}
With the establishment of substrate scope, we also wished to find some evidence to investigate the mechanism of this reaction. It was found that when running the reaction under the optimized reaction conditions depicted (Table 1, entry 7) in the presence of 3.0 equiv. of TEMPO ((2, 2, 6, 6-tetramethylpiperidin-1-yl)oxyl), barely no product was obtained even prolonged exposure to light. Besides, the PBN (α-phenyl N-tertiary-butyl nitrone) trapped CF3 radical adduct, using a solution of Umemoto's reagent in CH2Cl2, was detected by EPR experiment analysis under the irradiation of visible light in situ (see the Supporting information for more details) [17].
On the basis of the above observations and literature reports [13, 16, 18], a possible reaction mechanism was proposed (Scheme 3). After irradiation of Umemoto's reagent under visible light, CF3 radical was quickly released. Then a subsequent radical relay involving a HAT process resulted in the formation of a N-radical 1[8, 19, 21], which was followed by another HAT process to form C-radical 2[8, 20, 21]. The C-radical 2 could be quickly oxidized by excess Umemoto's reagent into oxocarbenium ion 3[8, 9], which finally fulfilled the reaction with purines to form the N-9 alkylated purine nucleoside derivatives 4.

|
Download:
|
| Scheme 3. Proposed mechanistic pathway. | |
{kind=link}
In summary, we have developed a novel and efficient method for the synthesis of purine nucleoside analogues from purine bases and a series of ethers. Using Umemoto's reagent under visible-light irradiation, the reaction of sp3 N-H in purines and sp3 C-H in alkyl ethers successfully provided the N-9 alkylated purine nucleoside derivatives in good yields. Compared with the previous approaches, the simplicity and novelty of this protocol as well as the excellent regioselectivity make this method particularly attractive.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21232002, 21572012) and Beijing Natural Science Foundation (No. 7152085).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.08.005.
| [1] |
(a) R. Garg, S. P. Gupta, H. Gao, et al., Chem. Rev. 99(1999) 3525-3602; (b) W. B. Parker, Chem. Rev. 109(2009) 2880-2893. |
| [2] |
(a) A. J. Wagstaff, D. Faulds, K. L. Goa, Drugs 47(1994) 153-205; (b) P. D. Welsby, J. Infect. 28(1994) 121-129; (c) A. D. Ruiter, R. N. Thin, Drugs 47(1994) 297-304; (d) W. A. Bowles, F. H. Schneider, L. R. R. Lewis, K. Robins, J. Med. Chem. 6(1963) 467-471; (e) R. B. Yan, Y. F. Wu, J. Liu, X. L. Zhang, X. S. Ye, Chin. Chem. Lett. 24(2013) 273-278. |
| [3] |
H. Rosemeyer, Chem. Biodivers. 1(2004) 361-401. DOI:10.1002/(ISSN)1612-1880 |
| [4] |
(a) R. J. Haslam, M. M. L. Davidson, J. V. Desjardins, Biochem. J. 176(1978) 83-95; (b) T. A. Rich, R. C. Shepard, S. T. Mosley, J. Clin Oncol. 22(2004) 2214-2232; (c) J. C. Martin, M. J. M. Hitchcock, E. De Clercq, W. H. Prusoff, Antiviral Res. 85(2010) 34-38; (d) E. De Clercq, Rev. Med. Virol. 19(2009) 287-299. |
| [5] |
(a) A. I. Votruba, J. Med Chem. 45(2002) 1918-1929; (b) U. Diederichsen, D. Weicherding, N. Diezemanna, Org. Biomol. Chem. 3(2005) 1058-1066; (c) G. A. Sheikha, P. L. Colla, A. G. Loi, Nucleosides Nucleotides Nucleic Acids 21(2002) 619-635. |
| [6] |
N.F. Zakirova, A.V. Shipitsyn, E.F. Belanovb, M.V. Jasko, Bioorg Med., Chem. Lett. 14(2004) 3357-3360. DOI:10.1016/j.bmcl.2003.12.107 |
| [7] |
(a) F. Vandendriessche, R. Snoeck, G. Janssen, J. Med. Chem. 35(1992) 1458-1465; (b) P. Franchetti, G. A. Sheikha, L. Cappellacci, J. Med. Chem. 38(1995) 4007-4013. |
| [8] |
H.M. Guo, C. Xia, H.Y. Niu, et al., Adv. Synth. Catal. 353(2011) 53-56. DOI:10.1002/adsc.201000682 |
| [9] |
I. Buslov, X. Hu, Adv. Synth. Catal. 356(2014) 3325-3330. DOI:10.1002/adsc.201400646 |
| [10] |
R. Huang, C.S. Xie, L. Huang, J.H. Liu, Tetrahedron 69(2013) 577-582. DOI:10.1016/j.tet.2012.11.020 |
| [11] |
(a) M. Ochiai, S. Yamane, M. Hoque, M. Saito, K. Miyamoto, Chem. Commun. 48(2012) 5280-5282; (b) L. He, J. Yu, J. Zhang, X. Q. Yu, Org. Lett. 9(2007) 2277-2280; (c) T. Fuchigami, T. Sato, T. Nonaka, J. Org. Chem. 51(1986) 427-432; (d) K. S. Feldman, M. M. Bruendl, K. Schildknegt, A. C. Bohnstedt, J. Org. Chem. 61(1996) 5440-5452; (e) H. Kim, J. Kim, S. Cho, S. Chang, J. Am. Chem. Soc. 133(2011) 16382-16385; (f) M. Ochiai, K. Miyamoto, T. Kaneaki, S. Hayashi, W. Nakanishi, Science 332(2011) 448-451; (g) A. P. Antonchick, R. Samanta, K. Kulikov, J. Lategahn, Angew. Chem Int. Ed. 50(2011) 8605-8608. |
| [12] |
(a) K. L. Skubi, T. R. Blum, T. P. Yoon, Chem. Rev. 116(2016) 10035-10074; (b) C. K. Prier, D. A. Rankic, D. W. C. MacMillan, Chem. Rev. 113(2013) 5322-5363; (c) N. A. Romero, D. A. Nicewicz, Chem. Rev. 116(2016) 10075-10166; (d) D. M. Arias-Rotondo, J. K. McCusker, Chem. Soc. Rev. 45(2016) 5803-5820. |
| [13] |
T. Umemoto, Chem. Rev. 96(1996) 1757-1778. DOI:10.1021/cr941149u |
| [14] |
B.P. Roberts, Chem. Soc. Rev. 28(1999) 25-35. DOI:10.1039/a804291h |
| [15] |
Y. Yasu, T. Koike, M. Akita, Angew. Chem. Int. Ed. 51(2012) 9567-9571. DOI:10.1002/anie.v51.38 |
| [16] |
(a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández, P. Melchiorre, Nat. Chem. 5(2013) 750-756; (b) S. Mattia, E. Arceo, I. D. Jurberg, C. Cassani, P. Melchiorre, J. Am. Chem. Soc. 137(2015) 6120-6123; (c) J. F. Franz, W. B. Kraus, K. Zeitler, Chem. Commun. 51(2015) 8280-8283; (d) M. Tobisu, T. Furukawa, N. Chatani, Chem. Lett. 42(2013) 1203-1205. |
| [17] |
X. Wang, Y. Ye, S. Zhang, et al., J. Am. Chem. Soc. 133(2011) 16410-16413. DOI:10.1021/ja207775a |
| [18] |
(a) C. P. Zhang, H. Wang, A. Klein, et al., J. Am. Chem. Soc. 135(2013) 8141-8144; (b) R. Z. Mao, D. C. Xiong, F. Guo, et al., Org. Chem. Front. 3(2016) 737-743. |
| [19] |
S.Z. Zard, Chem. Soc. Rev. 37(2008) 1603-1618. DOI:10.1039/b613443m |
| [20] |
Z. Cekovic, J. Serb. Chem. Soc. 70(2015) 287-318. |
| [21] |
(a) J. C. K. Chu, T. Rovis, Nature 539(2016) 272-275; (b) G. J. Choi, Q. L. Zhu, D. C. Miller, C. J. Gu, R. R. Knowles, Nature 539(2016) 268-271; (c) A. Noble, D. W. C. MacMillan, J. Am. Chem. Soc. 136(2014) 11602-11605. |