 2018, Vol. 29
2018, Vol. 29 



b The Institute of Seawater Desalination and Multipurpose Utilization, State Oceanic Administration, Tianjin 300192, China
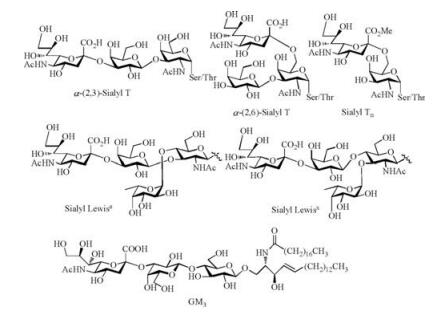
As a large family of natural nine-carbon sugar acids, sialic acid (Sia) is common in the higher animals and human beings. Nacetylneuraminic acid (Neu5Ac), N-glycolylneuraminic acid (Neu5Gc) and ketodeoxynonulosonic acid (KDN) are the most typical sialic acids. Neu5Ac is often linked to galactose or N-acetylgalactosamine by α-(2, 3)-, α-(2, 6)-glycosidic bonds, and is also found to form α-(2, 8)-, α-(2, 9)-disialyl structures. On the surface of cell membrane, sialic acid is generally associated with glycolipids and glycoproteins, which plays important roles in many biological processes, such as cell adhesion, antigen recognition, tumor migration and so on [1-3]. For instance, Neu5Ac was present at the terminal position of many tumor-associated carbohydrate antigens (TACAs), such as α-(2, 3)-sialyl T, α-(2, 6)-sialyl T, sialyl Tn, sialyl Lewisa/x, GM3 etc. (Scheme 1) [4, 5].

|
Download:
|
| Scheme 1. Chemical structures of some typical TACAs. | |
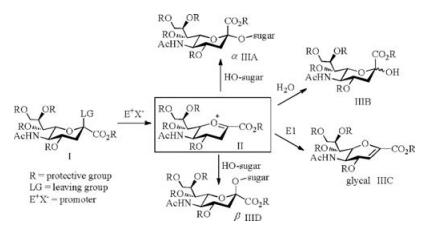
The sialylation reaction is the key step to the construction of glycoconjugates bearing sialic acids. To achieve an efficient sialylation with high yield and α-selectivity is the important criteria to evaluate the glycosylation reaction [6]. Due to the destabilizing effect of the COOR group at C2 and the absence of an —OH group at C3 of a sialyl donor (Ⅰ), the stereochemistry of the sialylation is difficult to control (Scheme 2). The sugar oxocarbenium intermediate Ⅱ was attacked by acceptor (HO-sugar) to give the desired α-sialoside product (ⅢA) accompanying with more thermodynamically stable non-natural β-sialoside (ⅢD). Meanwhile, 2, 3-elimination and hydrolysis are the main competition reactions during the glycosylation of acceptor. Glycal (ⅢC) and ⅢB could be obtained as the by-products to decrease the reaction yield.

|
Download:
|
| Scheme 2. Proposed mechanism of the sialylation reaction | |
In the past decades [6-11], great progress has been made on the sialylation to improve the α-selectivity, including the design of sialyl donors such as the introducing of leaving group on C2, the application of different leaving (auxiliary) groups at C1 and C3, the utilization of protective group on C4/C5, the modification of glycerol side chain on C7/C9. In addition, 1, 5-lactam and 1, 7-lactone derivatives were fabricated as the sialyl acceptors, which can relatively increase the activity of the hydroxyl on C8.
Similarly, extensive efforts have also been made to create glycosidic linkages that connect other sugar units [12-15]. For example, N-glycosylation reactions of nucleobases with pentose derivatives to elaborate nucleosides have been developed intensively and classified into three principal types: (1) Lewis acid promoted fusion reaction of peracylated sugars and heterocyclic rings [16, 17]; (2) Condensation of metal salts of heterocyclic systems with protected sugar halides [18, 19]; (3) The Hilbert-Johnson reaction [20-22]. In addition, to synthesize fucosides with high yields and α-stereoselectivities, various methodologies for fucosylation using glycoyl halide [23-26], trichloroacetimidate [27-29], acetate [30], sulfoxide [31], phosphate [32] and thioglycoside [33-39] as glycosyl donors were reported. However, the ideal glycosylation strategy with high generality and versatility suitable for most kinds of sugars was limited.
To address the extreme challenges raised in the synthesis of various complex glycoconjugates, here we provide a mini-review on the glycosylation reactions preactivated by (p-Tol)2SO/Tf2O pair, which was found to be highly efficient for various O-, C-sialylations and general O-, N-glycosyaltions. The glycosylation stereoselectivity and reactivity can be remarkably modulated by (p-Tol)2SO as additive.
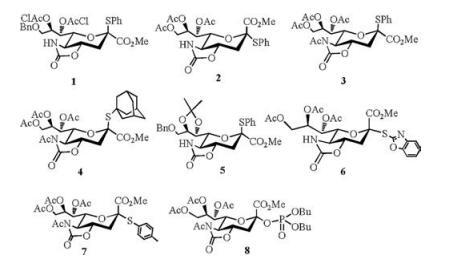
2. O-sialylation 2.1. 5-N, 4-O-carbonyl-protected 2-thio-sialoside as donorTo improve the α-selectivity outcome of the glycosylation, 5-N, 4-O-oxazolidinone trans-fused cyclic protecting group, as a powerful α-stereodirecting group, has been introduced to the various sialyl donors 1-8 (Fig. 1) [40-48]. Crich and co-workers [49] found that oxazolidinone ring has a larger dipole moment to retard the buildup of positive charge at the anomeric center, and consequently destabilize the oxocarbenium intermediates to reduce the anomeric effect, i.e., to favor the formation of equatorial glycoside.

|
Download:
|
| Fig. 1. The chemical structures of 5-N, 4-O-carbonyl-protected sialyl donors. | |
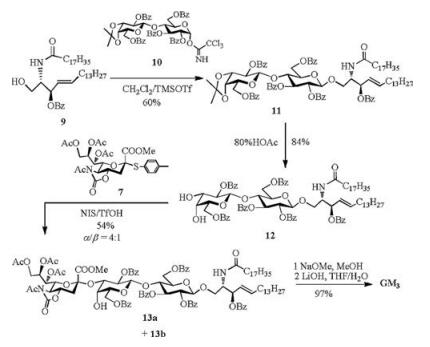
Thioglycoside is used as a kind of typical glycosyl donor in the synthesis of various oligosaccharides [50-52], since it has high chemical stability and can be readily activated in a glycosylation reaction. To access odorless or low odor thioglycoside is crucial for the operation of glycosylations. Considering that solid p-methylbenzenethiol is lower volatile than liquid thiophenol, Xing and coworkers [46] synthesized N-acetyl-5-N, 4-O-carbonyl-protected p-toluene 2-thio-sialoside (7). With NIS/TfOH as promotor in CH2Cl2-CH3CN mixed solvent at —40 ℃, 7 was successfully applied to various α-stereoselective sialylations with primary, secondary and tertiary alcohol as receptors [46, 53, 54]. Especially, for the synthesis of GM3 (α-Neu5Ac-(2, 3)-β-Gal-(1, 4)-β-Glc-(1, 1)-Cer) [4, 5, 55], thioglycoside 7 was employed in the key step in the presence of NIS/TfOH to build the sialyl α-(2, 3)-glycosidic linkage in trisaccharide 13 (54%, α/β = 4:1), and GM3 was successfully obtained from the building blocks (7, 9, 10) derived from Neu5Ac, lactose and L-serine in five linear steps with 26% overall yield (Scheme 3) [54].

|
Download:
|
| Scheme 3. Linear synthetic route for ganglioside GM3. | |
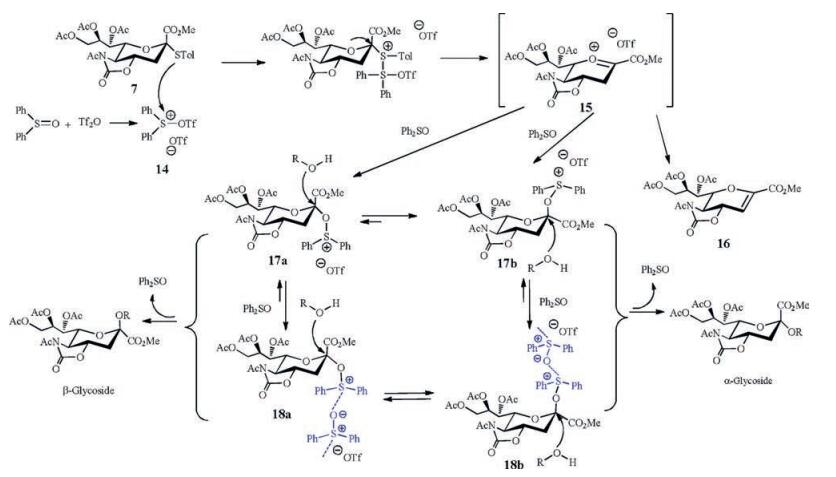
Ph2SO/Tf2O promotor system, which was firstly reported by van der Marel and co-workers [56], was capable of efficiently activating disarmed thioglycosides. Moreover, Ph2SO/Tf2O promotion protocol was a preactivation strategy in the glycochemical synthesis including one-pot glycosylation [57, 58]. The replacement of NIS/TfOH with Ph2SO/Tf2O as promotor was feasible for the sialylation reactions. Most importantly, Ph2SO was found as a powerful additive to significantly modulate the glycosylation activity and stereo-selectivity [59]. For example, when the amount of Ph2SO was increased from 2.0 equiv. to 5.0 equiv. in the sialylation of acceptor methyl 2, 3, 4-tri-O-benzyl-α-D-glucopyranoside with donor 7, the stereoselectivity (α/β) of the glycosylation remarkably dropped down from 12.6:1 to 1.6:1, and the corresponding product yield increased from 79% to ~100%. The similar results were obtained in the coupling of donor 7 with other acceptors. Considering both the yield and α-stereoselectivity of the sialylation, a compromised amount of Ph2SO (2.0-3.0 equiv.) was suggested to be used for the glycosylation reaction.
A hypothetical mechanism was proposed to interpret the unique functions of Ph2SO in the sialylation reaction (Scheme 4). There are three crucial reactive species: oxacarbenium ion 15, sialyloxosulfonium salts 17 and sialyloxosulfonium supramers 18. According to the study of Crich and co-workers [60], under the conditions without a suitable nucleophile the intermediate 15 could decompose to give glycal 16. An excessive Ph2SO (2-3 equiv.) would stabilize 15 to afford new intermediate C2-α/β-sialyloxosulfonium salt 17a/17b. The more reactive β-oxosulfonium species 17b could be attacked by an acceptor (ROH) to generate α-sialoside as the major product. There were many unprecedented results during the sialylation studies, and Kononov and co-workers [61, 62] put forward a novel concept "supramer approach" to explain these unexpected glycosylation outcomes. We speculate that largely excessive Ph2SO (>3.0 equiv.) could achieve C2-α/β-sialyloxosulfonium supramer 18a/18b, which were assembled from Ph2SO and 17a/17b through intermolecular electrostatic interactions. The supramer 18a/18b should possess similar reactivity, and be more stable than its corresponding precursor 17a/17b. Therefore, equilibration between 18a and 18b in the presence of Ph2SO (5.0 equiv.) gave a poor α-stereoselectivity of the sialylation, but excellent reaction yield (~100%) without glycal byproduct [59].

|
Download:
|
| Scheme 4. A proposed mechanism for the sialylation with Ph2SO/Tf2O as promotor. | |
To deeply compare the glycosylation profile of α-/β-oriented (N-acetyl)-5-N, 4-O-carbonyl-protected p-toluenethiosialosides donors, Xing and co-workers [63] prepared the analogues of 7 [46] including 19 [64], 20 [65], 21 [66], and systematically studied the sialylations promoted by NIS/TfOH and Ar2SO/Tf2O (Table 1). NIS/TfOH was shown to be a more efficient promotion system than (p-Tol)2SO/Tf2O to provide higher α-selectivities and reaction yields. In general, complex sialylation profiles were observed for N-deacetylated sialyl donors (19 and 20), which were more sensitive to the structures of acceptors. As a comparison, similar sialylation outcomes were afforded for N-acetylated donors (7 and 21).
|
|
Table 1 Sialylations of thiosialoside donors with different alcohol acceptors. |
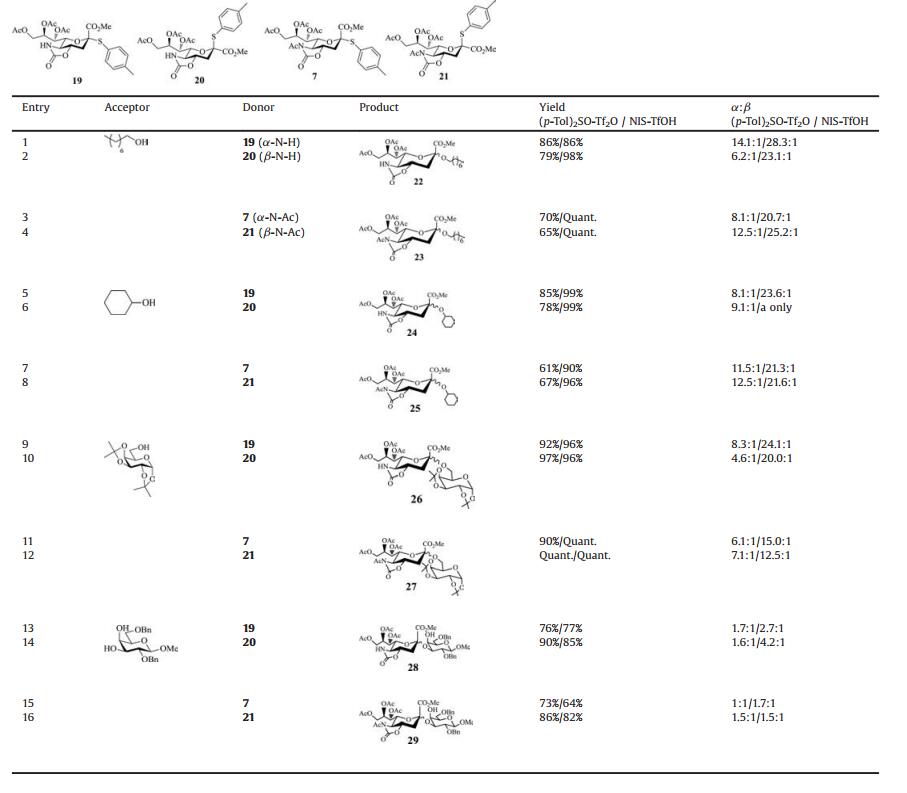
Except for NIS/TfOH and Ar2SO/Tf2O as the activator system for the thiosialoside, p-TolSCl/AgOTf [66] was also reported as a preactivation protocol, which had potential application in the one-pot iterative sialylconjugate synthesis [67].
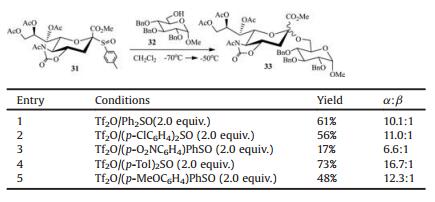
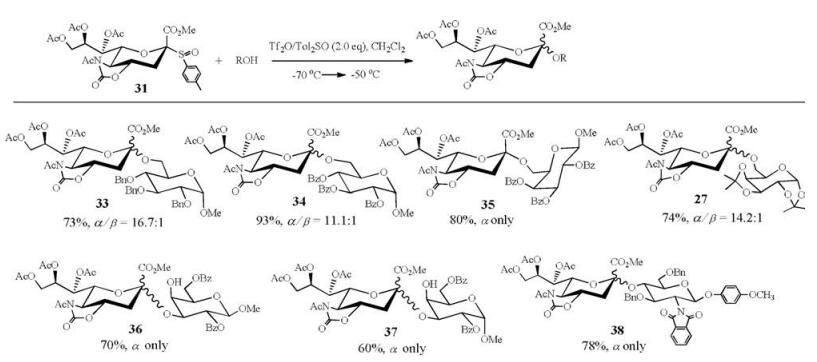
2.2. 5-N, 4-O-carbonyl-protected sialyl sulfoxide as donorGlycosyl sulfoxides have been considered as distinctive donors for chemical glycosylation with unreactive acceptors [68-72]. The attempts to obtain the sulfoxide of traditional N-acetyl p-toluene 2-thio-sialoside 30 was not successful. To our delight, N-Acetyl-5-N, 4-O-carbonyl-protected sialyl sulfoxide (31) was prepared by oxidization of donor 7 with m-CPBA [73] (Fig. 2). It is noted that (p-Tol)2SO was the best additive for the (Ar)2SO/Tf2O preactivation system (Table 2), since (p-Tol)2SO was the most matched sulfoxide for donor 31 with p-tolyl sulfoxide as leaving group. Sialyl sulfoxide 31 was shown to be quite efficient to synthesize α-(2, 3)-, α-(2, 4)-, and α-(2, 6)-sialosides (Scheme 5), which was better than its counterpart 7 in the syntheses of sialosides 33 and 36 [46, 53, 54].

|
Download:
|
| Fig. 2. The chemical structures of sialyl donors. | |
|
|
Table 2 Influence of different (Ar)2SO additive on the sialylation. |

|
Download:
|
| Scheme 5. Sialylation products obtained with sulfoxide 31 as donor. | |
3. C-sialylation
Owing to the formation of C—C bond in quaternary anomeric carbon, it is more difficult to prepare C-sialosides than other C-glycosides. In 1991, three research groups reported the first syntheses of simple allyl and hydroxymethyl C-sialosides [74-76]. However, the reaction yield or α-stereoselectivity was unsatisfatory. During the past decades, the SmI2-promoted Reformatskytype coupling reaction was developed to synthesize various C-sialosides. The reaction was usually carried out in anhydrous tetrahydrofuran. Sialyl sulfones, chlorides, sulfides and acetates were employed as the Neu5Ac donors in the C-glycosylation, and HMPA (hexamethylphosphoramide) was utilized as a ligand to accelerate the reaction [77-85].
Recently, N-acetyl-5-N, 4-O-carbonyl-protected sialyl phosphates (8) was acted as an efficient sialyl donor for the preparation of α-C-sialosides promoted by TMSOTf (trimethylsilyl trifluoromethanesulfonate) [64]. The author's group found that sialyl donor 7, which was the precursor of 8, was also a practical C-sialyl donor preactivated by (p-Tol)2SO/Tf2O with silyl enol ethers as acceptors to provide C-sialosides in high α-stereoselectivity and good product yield [86]. In the sialylation of acceptor acetophenone trimethylsilyl enol ether, (p-Tol)2SO could modulate the reaction yield from 61% to 84% with increasing the (p-Tol)2SO amount from 0.6 equiv. to 3.0 equiv. The results were simliar with the case using Ph2O as modulator in O-sialylation. Specifically, (p-Tol)2SO could stabilize the sugar oxacarbenium ion 15 to form sialyloxosulfonium salts 39 and sialyloxosulfonium supramer 40 (Scheme 6). However, when further increasing the (p-Tol)2SO amount to 4.0 equiv., the yield of C-sialoside 41 sharply declined to 15%. The result was different from the O-sialylation that 4.0 equiv. of Ph2O could still lead to the enhancement of the reaction yield. The sialyloxosulfonium intermediates were suggested to possess following reactivity order: 39b > 39a > 40a 

|
Download:
|
| Scheme 6. The crucial intermediates in the sialylation of donor 7 promoted by (p-Tol)2SO/Tf2O. | |
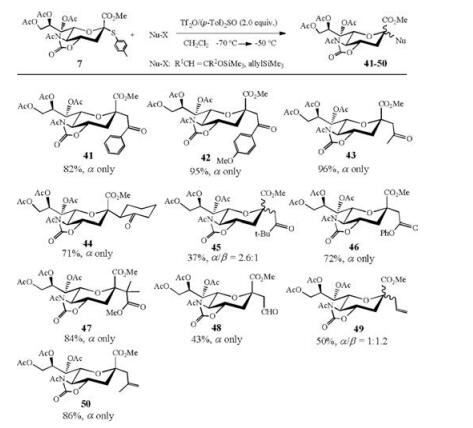
With thiosialoside 7 as donor and trimethylsilyl enol ethers and allyltrimethylsilanes as acceptors, a series of C-sialosides were successfully synthesized (Scheme 7). The π-nucleophiles used in Scheme 7 have different nucleophilicity values (N = 1.8-9.0) according to the definition of Mayr and co-workers [87]. It is noted that the C-sialylation efficiency is not only modulated by the added amount of (p-Tol)2SO, but also greatly influenced by the nucleophile reactivity. Strong π-nucleophiles with large nucleophilicity (N = 4.4-9.0) gave C-sialosides (41-44, 46-48, 50) in high α-stereoselectivity and high reaction yield [86].

|
Download:
|
| Scheme 7. C-sialylation products of donor 7 with different trimethylsilyl enol ethers and allyltrimethylsilanes. | |
4. N-Glycosylation
The synthesis of nucleosides and their analogues has become increasingly important for their broad utility in expanding genetic code [88], developing therapeutic agents [89-92], creating biological probes [93-95] and so on. In the past decades, based on the silyl-Hilbert-Johnson strategy, numerous glycosyl donors, such as 1-O-acetyl sugars, glycosyl trifluoroacetimidates, glycosyl ortho-alkynylbenzoates, thioglycosides, ribofuranosyl n-pentenyl orthoesters, 1-fluorofuranose, have been successfully employed for N-glycosylation of silylated pyrimidine and purine bases [96-101]. In addition, in most literatures, the reaction substrate was mainly limited to pyrimidine nucleobases or pentose sugars. For example, Tanaka and co-workers [99] reported the N-glycosylation of pyrimidines with thioglycosides as donors using several activators.
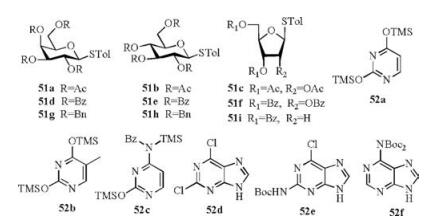
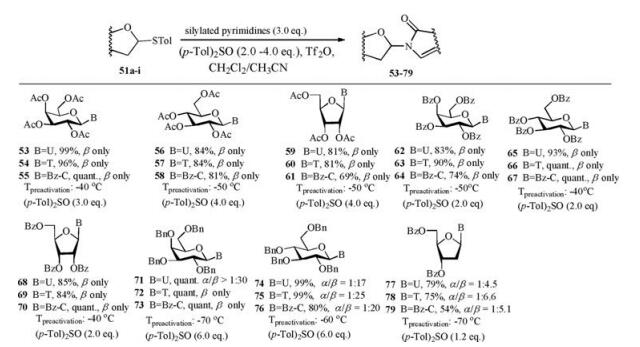
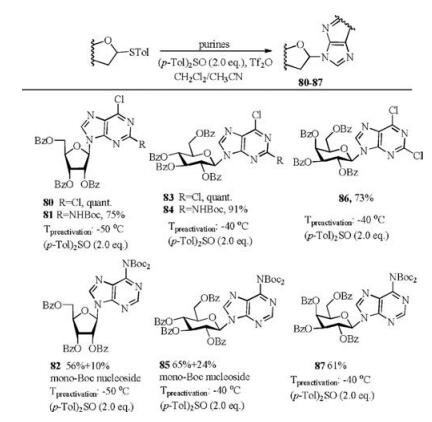
(p-Tol)2SO/Tf2O promoter was further applied in N-glycosylation reactions [102]. In order to establish one general strategy for O-, C- and N-glycosylation, the author's group performed the glycosylations of pyrimidines and purines with armed/disarmed sugar donors systematically (Fig. 3). Based on the powerful (p-Tol)2SO/Tf2O preactivation strategy, 35 nucleosides (53-87) were synthesized in high yields with high β-stereoselectivities (Schemes 8 and 9). We found that the reactivity of different donors was varied with the protecting groups and sugars, which was similar to the previous reports [103]. For example, to preactivate the donor completely, the disarmed acylated or benzoylated thioglycoside donors needed higher preactivation temperature than the corresponding perbenzyl protected donors. Moreover, we found that the yields and stereoselectivities of N-glycosylation were also significantly dependent upon the amount of (p-Tol)2SO additive (Table 3), which was closely related to the stability of the oxocarbenium intermediate cations formed after C—S bond cleavage. Larger (p-Tol)2SO amount was helpful to stabilize the active intermediates derived from donors and then increase the yields of N-glycosylation in most cases. However, the affection of the amount of (p-Tol)2SO additive to the stereoselectivity of N-glycosylation was more complexed and excellent β-stereoselectivity could be obtained by increasing (Table 3, entries 4-11) or decreasing (Table 3, entry 12-16) the amount of (p-Tol)2SO even without the anchimeric assistance at the C2 position of glycosyl donors (51g, 51h, 51i).

|
Download:
|
| Fig. 3. Structures of glycosyl donors and acceptors for nucleosides syntheses. | |

|
Download:
|
| Scheme 8. N-Glycosylation of pyrimidines with different glycosyl donors. | |

|
Download:
|
| Scheme 9. N-Glycosylation of purines with different glycosyl donors. | |
|
|
Table 3 Influence of different (p-Tol)2SO amount on the N-glycosylation. |
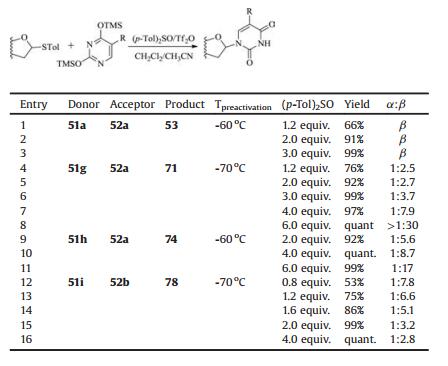
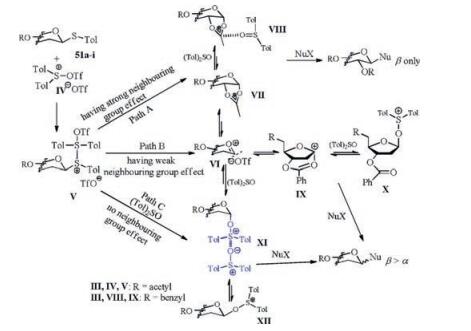
Then a plausible mechanism can be proposed by summarizing the results observed in the N-glycosylation (Scheme 10). In common, the diaryl sulfoxide bi(triflate) intermediate Ⅳ [104], in situ generated from (p-Tol)2SO and Tf2O, was attacked by the sulfur atom of the thioglycosides (51a-i) to give the sulfonium ion Ⅴ. Then the C—S bond of Ⅴ is cleaved to form the equilibria of Ⅵ/Ⅶ/Ⅷ, Ⅵ/Ⅸ/Ⅹ or Ⅵ/Ⅺ/Ⅻ, respectively. Detailedly, in the case of acyl protected sugars (Path A), the intermediate Ⅶ was produced due to the directing group at the C2 position. Ⅶ may be further stabilized by (p-Tol)2SO to form complex Ⅷ. As a consequnce, an efficient N-glycosylation was obatined in high yield and β stereoselectivity, which was consistent with the experimental observations (Table 3, entries 1-3). Similarly, the 3-O-benzoyl group in 51i seemed to participate in a remote anchimeric assistance to form an unstable intermediate Ⅸ [105, 106] which can be assaulted by excess (p-Tol)2SO (Path B). Then the mixture of Ⅸ and X provide an α/β anomeric mixture (Table 3, entries 12-16). In addition, when the neighbouring group effect did not exist (Path C), we speculated that the crucial intermediate XI formed as 18 and 40 was poised for the β-stereoselective coupling to the nucleobases (Table 3, entries 4-11).

|
Download:
|
| Scheme 10. A plausible mechanism of the N-glycosylation using (p-Tol)2SO/Tf2O preactivation protocol. | |
5. O-Fucosylation
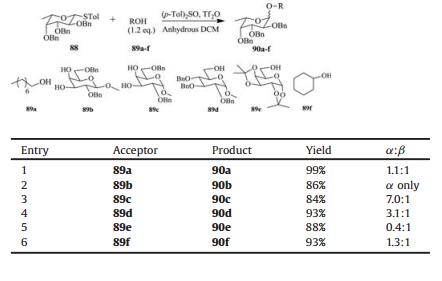
O-Fucosylation plays an important role in the synthesis of Lewis antigens. Unfortunately, it is still difficult to get high yields and excellent α-stereoselectivities for O-fucosylation. We have investigated the optimized conditions of O-fucosylation promoted by (p-Tol)2SO/Tf2O with perbenzylated fucosyl thioglycoside 88 as donor and octanol (89a) as acceptor [107]. At the preactivation temperature —70 ℃, the proportion of α-configuration product 90a was increased from 0.3:1 to 1.1:1 with increasing the amount of (p-Tol)2SO. When the preactivation temperature was increased from —70 ℃ to —60 ℃, the isolated product yield increased significantly from 65% to 99%, while the stereoselectivity of the glycosylation maintained 1.1:1 (α/β). Table 4 showed that high α-selectivity could be obtained with secondary sugar alcohols, and low α-selectivity was observed with simple non-sugar alcohols as acceptors.
|
|
Table 4 Fucosylation of 89a-f with 88 as glycosyl donor (6.0 eq. (p-Tol)2SO, —60 ℃). |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
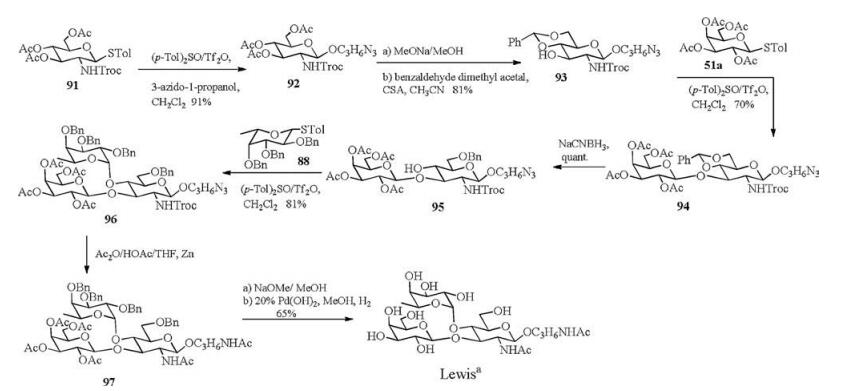
Under the optimized conditions for O-fucosylation, we successfully synthesized Lewisa containing L-fucose residue in 9 linear steps with a total 27% yield (Scheme 11). The (p-Tol)2SO/Tf2O preactivation strategy was used in all the glycosylation reactions in high yields and with excellent α-/β-stereoselectivities. Our O-fucosylation protocol should be meaningful for the preparation of other Lewis antigens, which could be further utilized for the cancer-related studies.

|
Download:
|
| Scheme 11. Synthetic route of Lewisa. | |
{kind=link}
6. Special application
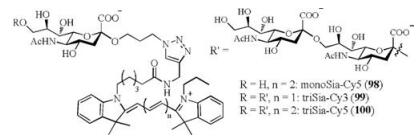
As the indispensable members of sugar epitope family, α-(2, 9)-di/oligosialic acids have been found in some kinds of cancer cells, such as human teratocarcinoma and mouse neuroblastoma cells [108, 109]. In addition to the functional glycoconjugates, there are various Sia-binding immunoglobulin-like lectins (Siglecs) that coexist as the receptors on the cell surface. The non-covalent interaction between aforementioned host and guest has been proved to be the immunization-related behavior in the organisms [110-114]. In 2014, Gao and coworkers elucidated the molecular basis of Sia recognition for the first time through solving the crystal structures of the paired immunoglobulin-like type Ⅱ receptors (PILRα and PILRβ) bearing Siglecs fold [115]. Recently, we designed and synthesized the fluorescently-labeled α-(2, 9)-trisialic acids (triSias, 99-100) with cyanine dyes (Fig. 4) to achieve the imaging of glycan-binding receptors, so as to establish an effective method to facilitate their visualization and further assessment [116].

|
Download:
|
| Fig. 4. The chemical structures of cyanine dye-tagged α-(2, 9)-mono/oligosialic acids 98-100. | |
{kind=link}
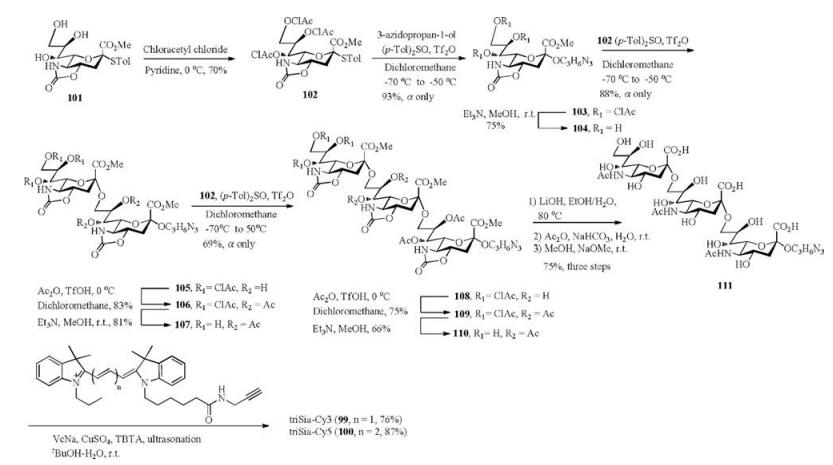
At the first stage, we prepared the azido-functionalized α-(2, 9)-trisialic acid 111 in 12 steps with 10% overall yield (Scheme 12). Both the introduction of azido group and the elongation of sugar chain were based on the preactivation protocol with (p-Tol)2SO/Tf2O as the promoter. Three α-glycosidic linkages were exclusively and efficiently constructed in 93%, 88% and 69% yield, successively. The final global deprotection of all the protective groups in trisaccharide 108 guaranteed the excellent water-solubility of the glycoconjugate. Then the triSia 111 was labeled by cyanine dyes (Cy3 or Cy5) with alkynyl group via Cu(Ⅰ)-catalyzed click reaction in the presence of ligand tris(benzyltriazolylmethyl)amine (TBTA), and the target compounds 99 and 100 were afforded in 76% and 87% yield, respectively.

|
Download:
|
| Scheme 12. Synthetic route of the Cy3/Cy5 -labeled triSias 99 and 100. | |
{kind=link}
To our delight, it was found that the triSia-Cy3 and triSia-Cy5 principally enriched on the membrane of PC-12 cells discretely because of the interaction between oligosaccharide and a certain kind of Siglec (Figs. 5a and b). However, monoSia-Cy5 (98) without net charge entered into the cytoplasm and gathered in the mitochondria within 1 min, which mainly exhibited the property of cyanine dye moiety (Fig. 5c). Furthermore, the spatial distribution information of the Siglecs was obtained if triSia-Cy3 and triSia-Cy5 were employed simultaneously to stain the cells. A pair of adjacent Siglec moieties probably bind with triSia molecules labeled by different dyes, and then the FRET process from Cy3 to Cy5 was triggered to export the region with higher spatial density of the Siglecs.

|
Download:
|
| Fig. 5. Confocal laser scanning microscopy images of PC-12 cells after incubating with triSia-Cy3 (a), triSia-Cy5 (b), and monoSia-Cy5 (red line) /MitoTracker Green (green line) (c). | |
{kind=link}
7. Conclusion and outlook
In summary, we have shown that the glycosylation based on (p-Tol)2SO/Tf2O preactivition strategy using thioglycosides or sialyl sulfoxide as donors is one of the most effective methods for preparation of glycoconjugates. Using this general glycosylation strategy, various O-, C-, and N-glycoconjugates, including related potential biological activity compounds antigen Lewisa, nucleosides, α-(2, 9)-trisialic acid, were successfully synthesized. The reaction yields and stereoselectivities were found to be highly dependent upon the reaction conditions, especially the amount of (p-Tol)2SO additive and the preactivation temperature, suggesting that (p-Tol)2SO would be coupled with the oxocarbenium cation reversibly to stabilize the glycosylation active intermediate.
By comparing the data reported in the related literatures, some general rules can be formulated, which should be helpful in the synthesis of other new glycoconjugates using this strategy. (1) The disarmed acylated thioglycoside donors need much higher preactivation temperature to be activated completely than the reactive deoxythioglycosides and perbenzyl protected donors; (2) To increase the glycosylation yield, much more (p-Tol)2SO are needed to trap the less stable oxocarbenium ion intermediate for acetyl protected donors than benzoyl protected donors; (3) Long reaction time is favored for the acceptors with weak nucleophilicity; (4) The stereochemical outcome is mainly determined by the anchimeric effect in the presence of neighboring group and by the equilibrium of the α-/β-glycosyl oxosulfonium salt mixture formed by (p-Tol)2SO reacting with oxocarbenium ion intermediate in the absence of neighboring group.
On the basis of the mechanism discussed above, we believed that the yield and stereoselectiviy of glycosylations via (p-Tol)2SO/Tf2O preactivition strategy could be tunable to a satisfactory degree by changing the preactivation temperature and the amount of (p-Tol)2SO additive regardless of the donors and acceptors used, demonstrating the high generality and versatility of this method. Further investigation to extrapolate the present additive effect to other glycosylations using different kinds of donors and promoters shall be an interesting topic and helpful for developing more general methodology to synthesize complex glycoconjugates.
AcknowledgmentThe project was financially supported by the National Natural Science Foundation of China (No. 21272027).
| [1] |
R. Schauer, Sialic Acids:Chemistry, Metabolism and Function[M]. New York: Spring-Verlag, 1982.
|
| [2] |
F. Lehmann, E. Tiralongo, J. Tiralongo, Cell. Mol. Life Sci. 63(2006) 1331-1354. DOI:10.1007/s00018-005-5589-y |
| [3] |
J. Tirolongo, I. Martinez-Duncker, Sialobiology:Structure, Biosynthesis and Function[M]. Bentham Science Publishers, 2013.
|
| [4] |
M.M. Fuster, J.D. Esko, Nat. Rev. Cancer 5(2005) 526-542. DOI:10.1038/nrc1649 |
| [5] |
G.S. Chen, Prog. Chem. 22(2010) 1753-1759. |
| [6] |
G.J. Boons, A.V. Demchenko, Chem. Rev. 100(2000) 4539-4565. DOI:10.1021/cr990313g |
| [7] |
B.O. Fraser-Reid, K. Tatsuta, J. Thiem, Glycoscience:Chemistry and Chemical Biology[M]. 2nd ed.. New York: Spring-Verlag, 2008, pp. 1313-1360.
|
| [8] |
F.F. Laing, L. Chen, G.W. Xing, Chin. J. Org. Chem. 29(2009) 1317-1324. |
| [9] |
D.H. Ye, J.F. Wang, H. Liu, et al., Prog. Chem. 22(2010) 91-100. |
| [10] |
B. Sun, Curr. Org. Chem. 20(2016) 1465-1476. DOI:10.2174/138527282014160419234226 |
| [11] |
T.J. Boltje, T. Buskas, G.J. Boons, Nat. Chem. 1(2009) 611-622. DOI:10.1038/nchem.399 |
| [12] |
Y. Hu, K. Yu, L. L. Shi, et al., J. Am Chem. Soc. (2017), doi: http://dx.doi.org/10.1021/jacs.7b07020.
|
| [13] |
X. Xiao, Y. Zhao, P. Shu, et al., J. Am. Chem. Soc. 138(2016) 13402-13407. DOI:10.1021/jacs.6b08305 |
| [14] |
S. Medina, M.J. Harper, E.I. Balmond, et al., Org. Lett. 18(2016) 4222-4225. DOI:10.1021/acs.orglett.6b01962 |
| [15] |
H. Liu, J.X. Liao, Y. Hu, Y.H. Tu, J.S. Sun, Org. Lett. 18(2016) 1294-1297. DOI:10.1021/acs.orglett.6b00216 |
| [16] |
J. Im, J. Kim, S. Kim, B. Hahn, F. Toda, Tetrahedron Lett. 38(1997) 451-452. DOI:10.1016/S0040-4039(96)02323-4 |
| [17] |
E. Diekmann, K. Friedrich, H.G. Fritz, J. Prakt. Chem. 335(1993) 415-424. DOI:10.1002/(ISSN)1521-3897 |
| [18] |
T. Ukita, H. Hayatsu, Y. Tomita, Chem. Pharm. Bull. 11(1963) 1068-1073. DOI:10.1248/cpb.11.1068 |
| [19] |
Z. Kazimierczuk, H.B. Cottam, G.R. Revankar, R.K. Robins, J. Am. Chem. Soc. 106(1984) 6379-6382. DOI:10.1021/ja00333a046 |
| [20] |
H. Vorbrueggen, Acc. Chem. Res. 28(1995) 509-520. DOI:10.1021/ar00060a007 |
| [21] |
L. Birkofer, A. Ritter, H.P. Kühlthau, Angew. Chem. 75(1963) 209-210. |
| [22] |
U. Niedballa, H. Vorbrueggen, J. Org. Chem. 39(1974) 3654-3660. DOI:10.1021/jo00939a008 |
| [23] |
S.S. Rana, J.J. Barlow, K.L. Matta, Carbohydr. Res. 96(1981) 231-239. DOI:10.1016/S0008-6215(00)81873-X |
| [24] |
U. Spohr, R.U. Lemieux, Carbohydr. Res. 174(1988) 211-237. DOI:10.1016/0008-6215(88)85093-6 |
| [25] |
A. Asnani, F.I. Auzanneau, Carbohydr. Res. 338(2003) 1045-1054. DOI:10.1016/S0008-6215(03)00053-3 |
| [26] |
D.K. Watt, D.J. Brasch, D.S. Larsen, L.D. Melton, J. Simpson, Carbohydr. Res. 325(2000) 300-312. DOI:10.1016/S0008-6215(00)00017-3 |
| [27] |
P. Zhang, K. Ng, C.C. Ling, Org. Biomol. Chem. 8(2010) 128-136. DOI:10.1039/B914193F |
| [28] |
J.M. Pletcher, F.E. McDonald, Org. Lett. 7(2005) 4749-4752. DOI:10.1021/ol051971s |
| [29] |
M.J.E. Silva, L. Cottier, R.M. Srivastava, D. Sinou, A. Thozet, Carbohydr. Res. 340(2005) 309-314. DOI:10.1016/j.carres.2004.11.016 |
| [30] |
M. Elofsson, S. Roy, L.A. Salvador, J. Kihlberg, Tetrahedron Lett. 37(1996) 7645-7648. DOI:10.1016/0040-4039(96)01702-9 |
| [31] |
L. Yan, D. Kahne, J. Am. Chem. Soc. 118(1996) 9239-9248. DOI:10.1021/ja9608555 |
| [32] |
O.J. Plante, E.R. Palmacci, R.B. Andrade, P.H. Seeberger, J. Am. Chem. Soc. 123(2001) 9545-9554. DOI:10.1021/ja016227r |
| [33] |
J. Xue, J. Zhu, R.E. Marchant, Z. Guo, Org. Lett. 7(2005) 3753-3756. DOI:10.1021/ol0514202 |
| [34] |
F. Yan, J. Xue, J. Zhu, R.E. Marchant, Z. Guo, Bioconjugate Chem. 16(2005) 90-96. DOI:10.1021/bc049805c |
| [35] |
A. Wang, F.-I. Auzanneau, Carbohydr. Res. 345(2010) 1216-1221. DOI:10.1016/j.carres.2010.03.038 |
| [36] |
M. Guillemineau, F.-I. Auzanneau, J. Org. Chem. 77(2012) 8864-8878. DOI:10.1021/jo301644w |
| [37] |
P.I. Abronina, A.I. Zinin, L.O. Kononov, Synlett. 26(2015) 2267-2271. DOI:10.1055/s-00000083 |
| [38] |
Y. Hua, G. Gu, Y. Du, Carbohydr. Res. 339(2004) 842-867. |
| [39] |
N. Ustyuzhanina, V. Krylov, A. Grachev, A. Gerbst, N. Nifantiev, Synthesis 23(2006) 4017-4031. |
| [40] |
H. Tanaka, Y. Nishiura, T. Takahashi, J. Am. Chem. Soc. 128(2006) 7124-7125. DOI:10.1021/ja0613613 |
| [41] |
M.D. Farris, C. De Meo, Tetrahedron Lett. 48(2007) 1225-1227. DOI:10.1016/j.tetlet.2006.12.061 |
| [42] |
D. Crich, W.J. Li, J. Org. Chem. 72(2007) 2387-2391. DOI:10.1021/jo062431r |
| [43] |
D. Crich, W.J. Li, J. Org. Chem. 72(2007) 7794-7797. DOI:10.1021/jo7012912 |
| [44] |
H. Tanaka, Y. Nishiura, T. Takahashi, J. Am. Chem. Soc. 130(2008) 17244-17245. DOI:10.1021/ja807482t |
| [45] |
B.N. Harris, P.P. Patel, C.P. Gobble, M.J. Stark, C. De Meo, Eur. J. Org. Chem(2011), 4023-4027. |
| [46] |
F.F. Liang, L. Chen, G.W. Xing, Synlett(2009), 425-428. |
| [47] |
C.H. Hsu, K.C. Chu, C.H. Wong, et al., Chem. Eur. J. 16(2010) 1754-1760. DOI:10.1002/chem.v16:6 |
| [48] |
K.C. Chu, C.T. Ren, C.Y. Wu, Angew. Chem. Int. Ed. 50(2011) 9391-9395. DOI:10.1002/anie.v50.40 |
| [49] |
P.K. Kancharla, C. Navuluri, D. Crich, Angew. Chem. Int. Ed. 51(2012) 11105-11109. DOI:10.1002/anie.201204400 |
| [50] |
G. Lian, X. Zhang, B. Yu, Carbohydr. Res. 403(2015) 13-22. DOI:10.1016/j.carres.2014.06.009 |
| [51] |
R.Z. Mao, F. Guo, D.C. Xiong, et al., Org. Lett. 17(2015) 5606-5609. DOI:10.1021/acs.orglett.5b02823 |
| [52] |
J.S. Huang, W. Huang, X. Meng, et al., Angew. Chem. Int. Ed. 54(2015) 10894-10898. DOI:10.1002/anie.201505176 |
| [53] |
L. Chen, F.F. Liang, M.F. Xu, G.W. Xing, Z.W. Deng, Acta Chim. Sinica 67(2009) 1355-1362. |
| [54] |
G.W. Xing, L. Chen, F.F. Liang, Eur. J. Org. Chem(2009), 5963-5970. |
| [55] |
R. Fuentes, R. Allman, M.D. Mason, Lung Cancer 18(1997) 21-33. |
| [56] |
J.D.C. Codee, R.E.J.N. Litjens, G.A. van der Marel, et al., Org. Lett. 5(2003) 1519-1522. DOI:10.1021/ol034312t |
| [57] |
L. Yang, Q. Qin, X.S. Ye, Asian J. Org. Chem. 2(2013) 30-49. DOI:10.1002/ajoc.201200136 |
| [58] |
Y.H. Wang, X.S. Ye, L.H. Zhang, Org. Biomol. Chem. 5(2007) 2189-2200. DOI:10.1039/b704586g |
| [59] |
Y.J. Wang, J. Jia, G.W. Xing, Carbohydr. Res. 346(2011) 1271-1276. DOI:10.1016/j.carres.2011.04.029 |
| [60] |
D. Crich, W. Li, Org. Lett. 8(2006) 959-962. DOI:10.1021/ol060030s |
| [61] |
L.O. Kononov, N.N. Malysheva, E.G. Kononova, A.V. Orlova, Eur. J. Org. Chem(2008), 3251-3255. |
| [62] |
L.O. Kononov, N.N. Malysheva, A.V. Orlova, Eur. J. Org. Chem(2009), 611-616. |
| [63] |
X.T. Zhang, Z.Y. Gu, G.W. Xing, Carbohydr. Res. 388(2014) 1-7. DOI:10.1016/j.carres.2014.02.006 |
| [64] |
A. Nobel, B. Delpech, D. Crich, Org. Lett. 14(2012) 1342-1345. DOI:10.1021/ol300255q |
| [65] |
C.C. Lin, N.P. Lin, C.C. Lin, et al., J. Org. Chem. 75(2010) 4921-4928. DOI:10.1021/jo100824s |
| [66] |
B. Sun, H. Jiang, Tetrahedron Lett. 52(2011) 6035-6038. DOI:10.1016/j.tetlet.2011.09.022 |
| [67] |
X.F. Huang, L.J. Huang, H.S. Wang, X.F. Ye, Angew. Chem. Int. Ed. 43(2004) 5221-5224. DOI:10.1002/(ISSN)1521-3773 |
| [68] |
J. Gildersleeve, R.A. Pascal, D. Kahne, J. Am. Chem. Soc. 120(1998) 5961-5969. DOI:10.1021/ja980827h |
| [69] |
J. Gildersleeve, A. Smith, K. Sakurai, S. Raghavan, D. Kahne, J. Am. Chem. Soc. 121(1999) 6176-6182. DOI:10.1021/ja990690a |
| [70] |
I. Alonso, N. Khiar, M. Martin-Lomas, Tetrahedron Lett. 37(1996) 1477-1480. DOI:10.1016/0040-4039(96)00043-3 |
| [71] |
D. Crich, S. Sun, J. Am. Chem. Soc. 119(1997) 11217-11223. DOI:10.1021/ja971239r |
| [72] |
D. Crich, S. Sun, J. Org. Chem. 62(1997) 1198-1199. DOI:10.1021/jo962345z |
| [73] |
Z.Y. Gu, J.X. Zhang, G.W. Xing, Chem. Asian J. 7(2012) 1524-1528. DOI:10.1002/asia.201200172 |
| [74] |
H. Paulsen, P. Matschulat, Liebigs Ann. Chem(1991), 487-495. |
| [75] |
K. Walliman, A. Vasella, Helv. Chim. Acta 74(1991) 1520-1532. DOI:10.1002/hlca.v74:7 |
| [76] |
J. Nagy, M. Bednarski, Tetrahedron Lett. 32(1991) 3953-3956. DOI:10.1016/0040-4039(91)80598-Z |
| [77] |
H.G. Bazin, Y. Du, T. Polat, R.J. Linhardt, J. Org. Chem. 64(1999) 7254-7259. DOI:10.1021/jo990564p |
| [78] |
B. Kuberan, S.A. Sikkander, H. Tomiyama, R.J. Linhardt, Angew. Chem. Int. Ed. 42(2003) 2073-2075. DOI:10.1002/anie.200351099 |
| [79] |
K.R. Dino, S.N. Baytas, R.J. Linhardt, et al., J. Org. Chem. 70(2005) 8197-8200. DOI:10.1021/jo050691n |
| [80] |
X. Yuan, D.K. Ress, R.J. Linhardt, J. Org. Chem. 72(2007) 3085-3088. DOI:10.1021/jo0623787 |
| [81] |
J.H. Kim, F. Huang, M. Ly, R.J. Linhardt, J. Org. Chem. 73(2008) 9497-9500. DOI:10.1021/jo801946y |
| [82] |
Z. Abdallah, G. Doisneau, J.M. Beau, Angew. Chem. Int. Ed. 42(2003) 5209-5212. DOI:10.1002/(ISSN)1521-3773 |
| [83] |
A. Malapelle, Z. Abdallah, G. Doisneau, J.M. Beau, Angew. Chem. Int. Ed. 45(2006) 6016-6020. DOI:10.1002/(ISSN)1521-3773 |
| [84] |
A. Malapelle, A. Coslovi, G. Doisneau, J.M. Beau, Eur. J. Org. Chem(2007), 3145-3157. |
| [85] |
A. Malapelle, Z. Abdallah, G. Doisneau, J.M. Beau, Heterocycles 77(2009) 1417-1424. DOI:10.3987/COM-08-S(F)118 |
| [86] |
Z.Y. Gu, X.T. Zhang, J.X. Zhang, G.W. Xing, Org. Biomol. Chem. 11(2013) 5017-5022. DOI:10.1039/c3ob40876k |
| [87] |
H. Mayr, B. Kempf, A.R. Ofial, Acc. Chem. Res. 36(2003) 66-77. DOI:10.1021/ar020094c |
| [88] |
I. Luyten, P. Herdewijn, Eur. J. Med. Chem. 33(1998) 515-576. DOI:10.1016/S0223-5234(98)80016-0 |
| [89] |
J. Stagg, M.J. Smyth, Oncogene 29(2010) 5346-5358. DOI:10.1038/onc.2010.292 |
| [90] |
E.D. Clercq, Med. Res. Rev. 30(2010) 667-707. |
| [91] |
A. Matsuda, T. Sasaki, Cancer Sci. 95(2004) 105-111. DOI:10.1111/cas.2004.95.issue-2 |
| [92] |
E.D. Clercq, Nat. Rev. Drug Discov. 6(2007) 1001-1018. DOI:10.1038/nrd2424 |
| [93] |
J.N. Wilson, E.T. Kool, Org. Biomol. Chem. 4(2006) 4265-4274. DOI:10.1039/b612284c |
| [94] |
A.P. Silverman, E.T. Kool, Chem. Rev. 106(2006) 3775-3789. DOI:10.1021/cr050057+ |
| [95] |
R.W. Sinkeldam, N.J. Greco, Y. Tor, Chem. Rev. 110(2010) 2579-2619. DOI:10.1021/cr900301e |
| [96] |
A. Sniady, M.W. Bedore, T.F. Jamison, Angew. Chem. Int. Ed. 50(2011) 2155-2158. DOI:10.1002/anie.v50.9 |
| [97] |
J.X. Liao, J. Sun, B. Yu, Tetrahedron Lett. 49(2008) 5036-5038. DOI:10.1016/j.tetlet.2008.06.042 |
| [98] |
Q.J. Zhang, J.S. Sun, Y.G. Zhu, F.Y. Zhang, B. Yu, Angew. Chem. Int. Ed. 50(2011) 4933-4936. DOI:10.1002/anie.v50.21 |
| [99] |
K. Mitsudo, W. Matsuda, S. Miyahara, H. Tanaka, Tetrahedron Lett. 47(2006) 5147-5150. DOI:10.1016/j.tetlet.2006.05.060 |
| [100] |
B. Fraser-Reid, P. Ganney, C.V.S. Ramamurty, A.M. Gómezb, J.C. Lópezb, Chem. Commun. 49(2013) 3251-3253. DOI:10.1039/c3cc41036f |
| [101] |
R. Noyori, M. Hayashi, Chem. Lett. 16(1987) 57-60. DOI:10.1246/cl.1987.57 |
| [102] |
G.J. Liu, X.T. Zhang, G.W. Xing, Chem. Commun. 51(2015) 12803-12806. DOI:10.1039/C5CC03617H |
| [103] |
Z. Zhang, I.R. Ollmann, C.H. Wong, et al., J. Am. Chem. Soc. 121(1999) 734-753. DOI:10.1021/ja982232s |
| [104] |
J.M. Haberman, D.Y. Gin, Org. Lett. 5(2003) 2539-2541. DOI:10.1021/ol034815z |
| [105] |
M. Kurosu, K. Li, J. Org. Chem. 73(2008) 9767-9770. DOI:10.1021/jo801408x |
| [106] |
F. Yang, Y.G. Zhu, B. Yu, Chem. Commun. 48(2012) 7097-7099. DOI:10.1039/c2cc33155a |
| [107] |
C.Y. Li, G.J. Liu, W. Du, Y. Zhang, G.W. Xing, Tetrahedron Lett. 58(2017) 2109-2112. DOI:10.1016/j.tetlet.2017.04.056 |
| [108] |
M.N. Fukuda, A. Dell, J.E. Oates, M. Fukuda, J. Biol. Chem. 260(1985) 6623-6631. |
| [109] |
S. Inoue, G.L. Poongodi, N. Suresh, H.J. Jennings, Y. Inoue, J. Biol. Chem. 278(2003) 8541-8546. DOI:10.1074/jbc.M212799200 |
| [110] |
P.R. Crocker, J.C. Paulson, A. Varki, Nat. Rev. Immunol. 7(2007) 255-266. DOI:10.1038/nri2056 |
| [111] |
C.D. Rillahan, M.S. Macauley, E. Schwartz, et al., Chem. Sci. 5(2014) 2398-2406. DOI:10.1039/c4sc00451e |
| [112] |
A. Varki, Nature 446(2007) 1023-1029. DOI:10.1038/nature05816 |
| [113] |
R. Schauer, Curr. Opin. Struct. Biol. 19(2009) 507-514. DOI:10.1016/j.sbi.2009.06.003 |
| [114] |
X. Chen, A. Varki, ACS Chem. Biol. 5(2010) 163-176. DOI:10.1021/cb900266r |
| [115] |
Q. Lu, G. Lu, J. Qi, et al., Proc. Natl. Acad. Sci. U. S. A. 111(2014) 8221-8226. DOI:10.1073/pnas.1320716111 |
| [116] |
X.T. Zhang, Z.Y. Gu, L. Liu, S. Wang, G.W. Xing, Chem. Commun. 51(2015) 8606-8609. DOI:10.1039/C5CC01907A |