 2017, Vol. 28
2017, Vol. 28 



b Key Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China
In recent years, worldwide energy shortage and environmental pollution aggravate gradually. Semiconductor photocatalysis, regarded as a green purification technology, has attracted extensive interest [1-3]. Many traditional semiconductor photocatalytic materials, such as titanium dioxide (TiO2), zinc oxide (ZnO) have been studied because of low cost and long-term stability [4-7]. However, the poor visible-light absorption and high recombination rate of photogenerated electrons and holes are major barriers that need to be surmounted, motivating the attempts for synthesis of highly efficient visible-light-driven photocatalysts [7, 8].
Layered bismuth-based photocatalysts, such as Sillén structured BiOX (X = Cl, Br, I), Aurivillius structured Bi2MO6 (M = W, Mo), have recently aroused more and more attention because of their excellent photo-oxidation ability and high stability [9-14]. Lately, several new layered bismuth-based photocatalytic materials, including Bi2O2[BO2(OH)], Bi2O2(OH)(NO3), MBiO2X (M = Sr, Ba; X = Cl, Br), LiBi3O4Cl2, Bi2MO4Cl (M = Eu, Gd), Bi4NbO8Br, BiOIO3 and Bi(IO3)3 have been developed by our group [15-23]. Among these materials, BiOIO3 possesses a noncentrosymmetric polar layered crystal structure composed of (Bi2O2)2+ layers and interbedded (IO3)- anions. The polarization electric field and electrostatic field can induce efficient separation of photoinduced charges carriers and thereby endow BiOIO3 with remarkable photocatalytic activity [24]. However, BiOIO3 is still limited by the dissatisfactory light utilization efficiency in visible region. Heterostructure fabrication as one of the most efficacious means for enhancing photoabsorption and accelerating photocatalytic activity attracts huge interest. Dong et al. and our group demonstrated that heterostructure fabrication by coupling BiOIO3 with Bi semimetal, BiOI, Bi2WO6, etc. can enhance the photocatalytic performance of BiOIO3 [25-27]. Meanwhile, silver halide semiconductor photocatalystsAgX (X = Br, I) are intensively investigated owing to the facile precipitation preparation and high visible light absorption [28]. Among the AgX group, AgI exhibits both high photosensitization ability and benign photocatalytic activity [29]. For instance, AgI/AgIO3 and AgI/Bi2WO6 photocatalyst show noteworthy visiblelight-responsive photocatalytic activity [30, 31]. Based on the structural benefits of BiOIO3 and optical advantages of AgI, fabrication of AgI/BiOIO3 heterostructure may be a feasible strategy to strengthen the photocatalytic performance of both components.
In this work, the AgI/BiOIO3 heterostructure photocatalysts are prepared by a facile in-situ crystallization route. The photocatalytic properties of as-obtained samples are studied by decomposition of model dye methyl orange (MO), phenols and antibiotics separately under ultraviolet (UV) light and visible light (λ > 420 nm) illumination. The results revealed that AgI/BiOIO3 exhibits much higher photocatalytic activity than AgI and BiOIO3 regardless of light source. The mineralization ability of AgI/BiOIO3 was also confirmed. In addition, the AgI/BiOIO3 heterostructure photocatalysts are characterized systematically via various techniques, and the different photocatalytic mechanisms under UV and visible light are investigated in detail (Scheme 1).

|
Download:
|
| Scheme 1. Schematic illustration of fabrication process for the AgI/BiOIO3 heterostructures. | |
{kind=link}
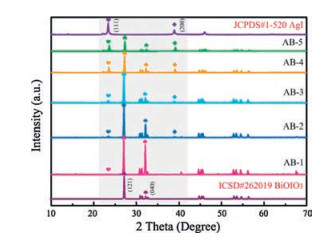
The structure and composition of AgI/BiOIO3 photocatalysts are characterized by XRD, XPS, SEM, TEM, HRTEM and EDXmapping. XRD patterns of all the samples including BiOIO3, AgI and AgI/BiOIO3 photocatalysts with different molar ratios are displayed in Fig. 1. The characteristic diffraction peaks of pristine BiOIO3 and AgI accord with the database of ICSD #262019 and JCPDS#1-520, respectively. With reference to the AgI/BiOIO3 samples, their XRD patterns contain the diffraction peaks of both pristine BiOIO3 and AgI, and the intensity of characteristic AgI (111) and (200) peaks apparently enhances with increasing the molar ratio of KI to BiOIO3. On the contrary, the (121) and (040) peaks of BiOIO3 continuously decrease from AB-1 to AB-5. Meantime, no other impurity peaks can be found. The above results revealed that the AgI/BiOIO3 composite photocatalysts have been successfully obtained and the crystalline phase and crystal structure are almost not changed compared to the two pristine samples.

|
Download:
|
| Fig. 1. XRD patterns for BiOIO3, AgI and AgI/BiOIO3 samples. | |
{kind=link}
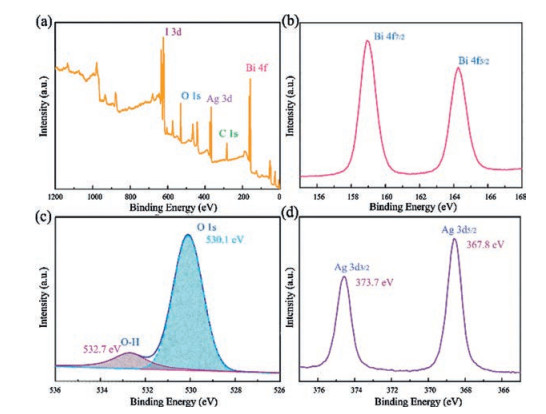
XPS is conducted to analyze the surface composition and chemical state of related elements of samples. As depicted in Fig. 2a, Bi, I, O, Ag and C elements can be detected in AB-3. The C 1s peak is resulted from the extraneous hydrocarbon of the XPS instrument. Fig. 2b displays the Bi 4f high-resolution XPS spectrum. The peaks at 159.0 eV and 164.4 eV originate from the Bi 4f7/2 and Bi 4f5/2 of Bi3+ ions, respectively [32-35]. The highresolution XPS spectrum of O 1 s is shown in Fig. 2c. The two peaks located at 530.1 eV and 532.7 eV can be attributed to the lattice O of BiOIO3 and the H2O molecules absorbed on the surface, respectively. Fig. 2d presents that the Ag 3d characteristic XPS spectrum of the AB-3 sample consists of two peaks at 367.8 eV and 373.7 eV, which match well with the feature of Ag+ 3d5/2 and Ag+ 3d3/2 [25]. Combined with the above XRD results, formation of the AgI/BiOIO3 photocatalysts was confirmed.

|
Download:
|
| Fig. 2. XPS survey spectra (a) and high-resolution XPS spectra for Bi 4f (b), O 1s (c) and Ag 3d (d) of AB-3. | |
{kind=link}
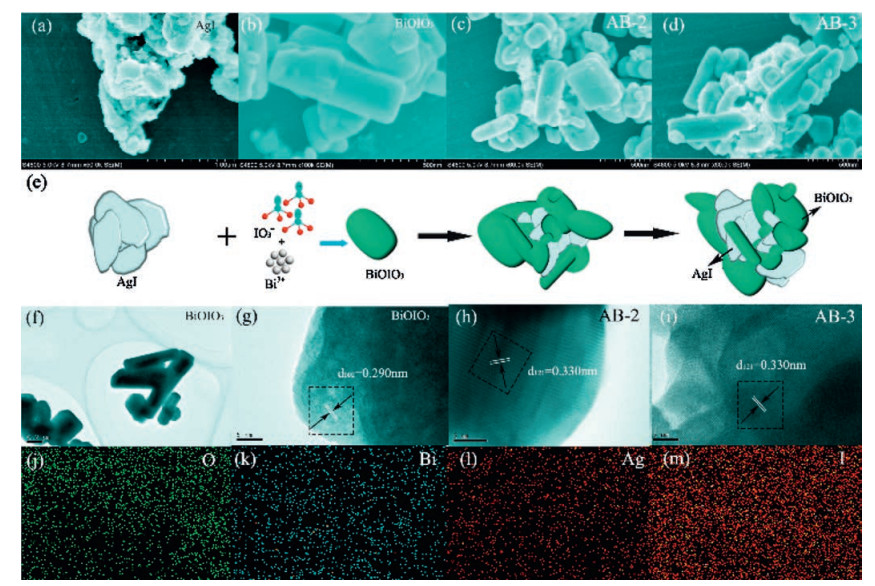
SEM, TEM, HRTEM and EDX-mapping are conducted to study the microstructure, morphology and element distribution of samples. As can be seen in Fig. 3a, AgI product consists of irregular nanoplates with size of ~50 nm. For BiOIO3, it displays flake structure and smooth surface with width of 0.2-1.0 μm and thickness of ~10 nm (Fig. 3b). Fig. 3c and d show the SEM image of AB-2 and AB-3, respectively. It can be found that AgI nanoplates were deposited on the surface of BiOIO3 flakes, and the morphology of BiOIO3 does not show obvious change. The schematic illustration of the formation process of AgI/BiOIO3 heterostructure is depicted in Figs. 3e. For further confirmation on the structure, the TEM image of BiOIO3 and HRTEM images of BiOIO3, AB-2 and AB-3 are shown in Fig. 3f–i. The lattice fringe with distance of 0.290 nm and 0.330 nm correspond to (002) and (121) planes of BiOIO3, respectively [36]. The lattice fringe of AgI is hard to be found due to its instability exposing to high-energy electron beam [25, 37]. The EDX-mapping of the AB-3 sample indicates that there is a uniform distribution of O, Bi, Ag and I elements (Figs. 3j–m). The above results provided solid evidence that AgI and BiOIO3 co-exist in the composite photocatalyst with close interaction.

|
Download:
|
| Fig. 3. SEM images for AgI (a), BiOIO3 (b), AB-2 (c), AB-3 (d). Schematic illustration for formation process of AgI/BiOIO3 composites (e). TEM (f) and HRTEM (g) images of BiOIO3 sample. TEM images of AB-2 (h) and AB-3 (i) samples.EDX mapping of AB-3 (j-m). | |
{kind=link}
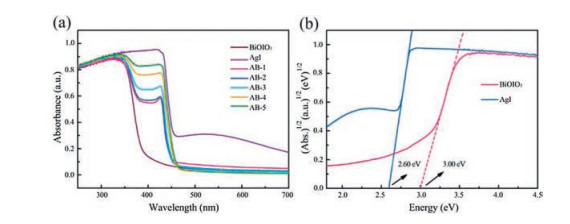
In Fig. 4a, the UV–vis DRS was used to evaluate the optical absorption properties of AgI, BiOIO3 and AgI/BiOIO3 composites. It can be seen that the optical absorption of BiOIO3 is mainly in UV region due to the absorption edge of pure BiOIO3 locating at around 400 nm, while the absorption band edge of AgI is approximately 470 nm. With the increase of AgI content, the absorption edge shows gradual red-shift to visible light region from AB-1 to AB-5, indicating that the responsive range of BiOIO3 in visible region can be remarkably extended. As revealed in Fig. 4b, the band gaps of the AgI and pure BiOIO3 are determined to be 2.6 eV and 3.0 eV, respectively.

|
Download:
|
| Fig. 4. UV-visdiffuse reflectance spectra for the BiOIO3, AgI and AgI/BiOIO3 composites (a). The determined band gaps of the BiOIO3 and AgI (b). | |
{kind=link}
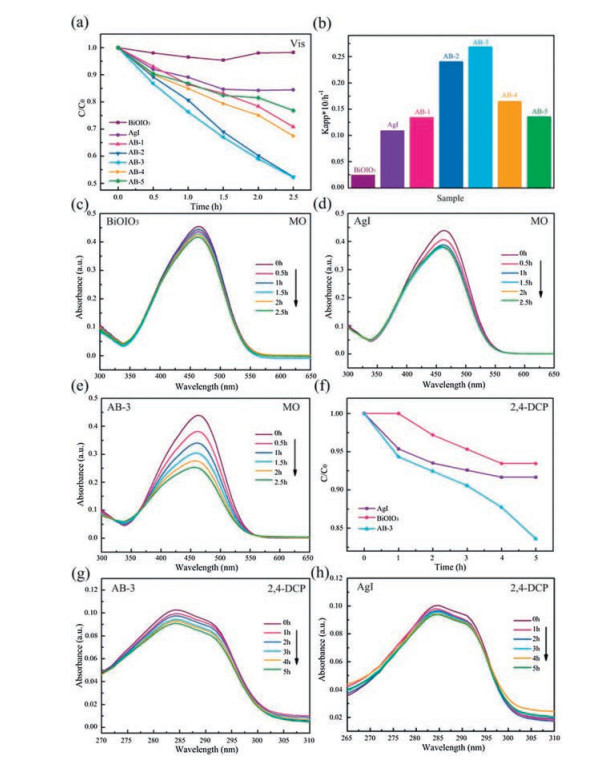
Considering that the pure BiOIO3 mainly responds to UV light, the photocatalytic performance of AgI, BiOIO3 and AgI/BiOIO3 composites is first evaluated by MO degradation experiments under UV light [28, 38]. Fig. 5a shows the photocatalytic degradation curves of MO. It can be concluded that the photocatalytic activities of all the AgI/BiOIO3 composites are higher than that of pure AgI and BiOIO3, and AB-3 can remove 94.7% of MO within 5 min UV-light illumination. As seen from Fig. 5b, the apparent rate constant for degrading MO over AB-3 is 0.572 min-1, which is 3.1 and 6.5 times higher than that of BiOIO3 (0.183 min-1) and AgI (0.088 min-1), respectively. As exhibited in Figs. 5c-f, the characteristic absorption band of MO over AgI, BiOIO3, AB-3 and AB-4 are gradually decreased with prolonging the irradiation time of UV light, implying the decomposition of MO.

|
Download:
|
| Fig. 5. Photocatalytic degradation curves of MO (a) and apparent rate constants for degrading MO (b) over the BiOIO3, AgI and AgI/BiOIO3 composite photocatalysts under UV light illumination. Temporal absorption spectra over BiOIO3(c), AgI (d), AB-3 (e) and AB-4 (f) samples. | |
{kind=link}
Fig. 6a shows the MO degradation capability of AgI, BiOIO3, and AgI/BiOIO3 under visible light (λ > 420 nm). It can be found that all the AgI/BiOIO3 composite samples exhibit remarkably enhanced photocatalytic activities compared to the two pristine samples. It should be noticed that the best photocatalytic performance is also observed for the AB-3, consistent with the above MO degradation result under UV-light. As depicted in Fig. 6b, the AB-3 photocatalyst has the highest photodegradation rate (0.269 h-1), which is 11.2 and 2.0 times higher than that of the pure BiOIO3 (0.024 h-1) and AgI (0.135 h-1) under visible light irradiation (λ > 420 nm). Figs. 6c–e demonstrate the instantaneous absorption spectra of MO over BiOIO3, AgI and AB-3. With the increase in irradiation time, the MO molecules are degraded gradually. Meantime, AB-3 shows the fastest decline, confirming the best photocatalytic performance.

|
Download:
|
| Fig. 6. 6. Photocatalytic degradation curves of MO (a) and apparent rate constants for degrading MO (b) over the BiOIO3, AgI and AgI/BiOIO3 composite photocatalysts under visible light illumination (λ > 420 nm). Temporal absorption spectra over BiOIO3(c), AgI(d) and AB-3 (e) samples. Photocatalyticdegradation curves of 2, 4-DCP over the pure BiOIO3, AgI and AB-3 composite photocatalysts under visible light illumination (f). Temporal absorption spectra of 2, 4-DCP over BiOIO3(g) and AgI(h). | |
{kind=link}
The enhanced visible-light photocatalytic activity of AB-3 was further confirmed by degrading 2, 4-dichlorophenol (2, 4-DCP). As exhibited in Fig. 6f, AB-3 possesses the highest photocatalytic activity compared with AgI and BiOIO3. With increasing the irradiation time, the characteristic absorption band of 2, 4-DCP gradually decreases (Figs. 6g–h). Combined the UV-light photodegradation results, it is safely concluded that the photocatalytic activity can be largely enhanced regardless of the light source.
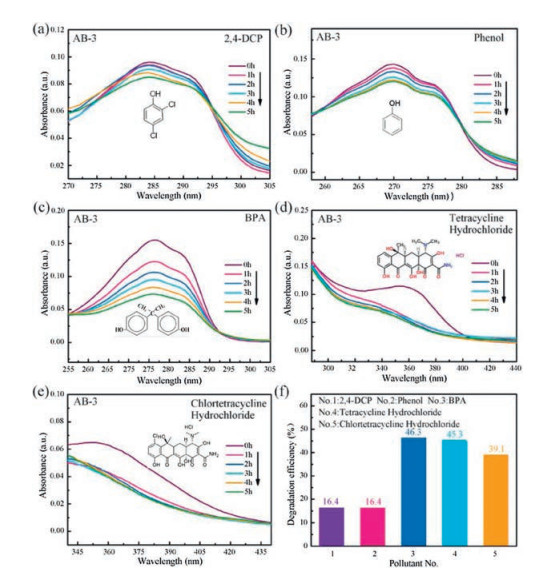
In order to further confirm the nonselective photocatalytic ability of AgI/BiOIO3 composites, the photocatalytic performance of AB-3 is evaluated by photo-decomposition of diverse stubborn contaminants, including colorless phenol, bisphenol A (BPA), pharmaceuticals tetracycline hydrochloride, and chlortetracycline hydrochloride under visible light irradiation (λ > 420nm). The temporal absorption spectra of the above pollutants over AB-3 are depicted in Figs. 7a-e. Their characteristic absorption bands are gradually reduced with increasing irradiation time, demonstrating the decomposition of these contaminants catalyzed by AB-3. To quantificationally assess the degradation performance, the removal efficiencies are showed in Fig. 7f, which are 16.4%, 16.4%, 46.3%, 45.3% and 39.1% for 2, 4-DCP, phenol, BPA, tetracycline hydrochloride and chlortetracycline hydrochloride within 5h illumination, respectively, implying the universal photocatalytic activity of AgI/BiOIO3.

|
Download:
|
| Fig. 7. Temporal absorption spectra of 2, 4-DCP (a), phenol (b), BPA (c), tetracycline hydrochloride (d) and chlortetracycline hydrochloride (e) over the AB-3 composite photocatalyst under visible light (λ > 420 nm) irradiation. Degradation efficiency of diverse contaminants under visible light irradiation (f). | |
{kind=link}
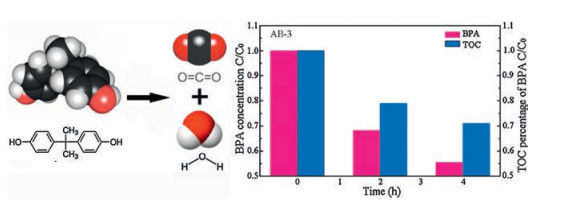
The total organic carbon (TOC) of BPA is determined to investigate the mineralization ability of AB-3. The TOC data over AB-3compositefor 0, 2 and 4h degradation are shownin Fig. 8.The photodegradation efficiency and mineralization ratio of organic carbon of BPA is 44.6% and 30%, respectively, after 4 h visible-light irradiation. It can be concluded that a majority of BPA is decomposed into CO2 and H2O. These evidences apparently demonstrated that AB-3 composite photocatalyst possesses the significantly enhanced visible-light photocatalytic activity and mineralization ability.

|
Download:
|
| Fig. 8. Photocatalytic degradation efficiency and TOC percentage of BPA over the AB-3 composite photocatalyst under visible light illumination. | |
{kind=link}
To further verify the photocatalytic stability of as-prepared sample, XRD of the sample after photoreaction was conducted. Compared with that of AB-3 before photoreaction, the XRD pattern after light irradiation has no obvious change (Fig. S1 in Supporting information). It demonstrates that the AB-3 has a good photocatalytic stability.
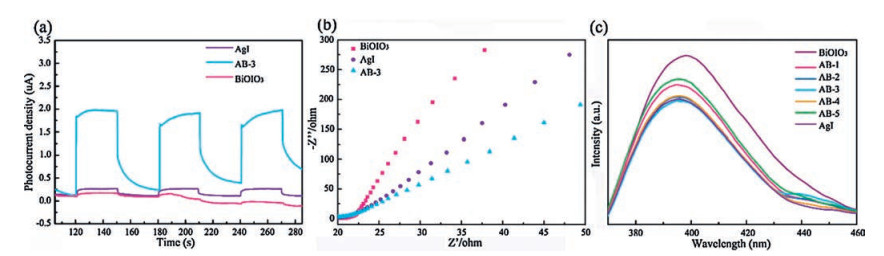
According to the above experimental results, the different photocatalytic activity enhancement mechanisms of the sample under UV and visible light irradiation are investigated in detail. The photocatalytic performance is mainly associated with the optical absorption and charge separation and transfer efficiencies [39]. Transient photocurrent of AgI, BiOIO3 and AB-3, as a valid technique, was employed to reveal the charge separation dynamics. As presented in Fig. 9a, AB-3 produces a drastically strengthened current density compared with AgI and BiOIO3 samples, indicating much more efficient charge separation. To explore the transfer behavior on the surface of the catalysts, electrochemical impedance spectra (EIS) are obtained. As seen from Fig. 9b, AB-3 composite shows a relatively smaller radius, demonstrating its larger interfacial charge transfer efficiency, as a small radius reflects a low charge transfer resistance. Photoluminescence spectroscopy (PL) is used to investigate the recombination rate of electrons and holes. It is stated that the high fluorescence intensity is mainly caused by high recombination of electrons and holes [40, 41]. As depicted in Fig. 9c, the emission peaks of AgI/BiOIO3 samples located at around 390 nm show markedly decreased intensities, implying that heterostructure fabrication plays an important role in hindering the charge recombination. It can be found that AB-3 exhibits the lowest emission intensity and the best photocatalytic performance, which is in accordance with pollutants degradation experiment. As depicted in Fig. S2 in Supporting information, BiOIO3 is a n-type semiconductor due to the positive slope of 1/C2 vs. V plot. Meantime, the flat band potential (Ffb) of BiOIO3 is calculated as -0.62 eV vs. saturated calomel electrode (SCE), which equals to -0.38 eV vs. normal hydrogen electrode (NHE). As the conduction band (CB) position is 0.1–0.3 eV higher than the flat potential, the CB of BiOIO3 is estimated to be -0.68 eV [42]. According to the DRS data, the valence band (VB) of BiOIO3 is 2.32 eV. Based on our previous reports, the CB potential of AgI that synthesized by the same method is about -0.15 eV [29]. Thus, the VB level of AgI is 2.45 eV according to its band gap (2.6 eV).

|
Download:
|
| Fig. 9. Comparison of transient photocurrent responses (a) and EIS Nynquist plots (b) for pure BiOIO3, AgI and AB-3 under UV-Vis light irradiation (0.1 mol/L Na2SO4). PL spectra of pure BiOIO3, AgI and AgI/BiOIO3 composites under the excitation of 245 nm (c). | |
{kind=link}
Based on the active species trapping experimental results (Figs. S3, S4 in Supporting information), the photocatalytic mechanism of the AgI/BiOIO3 under UV and visible light irradiation is separately deduced. Fig.S5a demonstrates the proposed mechanism with UV light illumination. Both AgI and BiOIO3 can be excited and produce electron-hole pairs. On account of the CB of AgI (-0.15 eV) is more positive than the CB of BiOIO3 (-0.68 eV), the photogenerated electrons on the CB of BiOIO3 would transfer to the CB of AgI. Meanwhile, the holes would transfer from the VB of AgI (2.45 eV) to that of BiOIO3 (2.32 eV) due to the more negative VB potential of BiOIO3. The holes and electrons can separately react with OH- and O2 to produce ·OH and ·O2-. As the potential of the VB of BiOIO3 (2.32 eV) is lower than the redox potential of ·OH/H2O (2.38 eV) [43], the oxidation ability of holes is insufficient. Therefore, the AgI/BiOIO3 heterostructure shows moderately improved photocatalytic activity under UV light irradiation. Fig.S5b shows the proposed photocatalytic mechanism of the AB-3 upon excitation with visible light. BiOIO3 almost cannot produce the electronhole pairs due to its wide band gap (3.0 eV). AgI can absorb visible light and be excited to generate electron-hole pairs. The electrons in AgI can reduce O2 to ·O2-, and holes oxidize OH- into ·OH radicals. In addition, the more negative VB potential of BiOIO3 favors the transfer of the holes from the VB of AgI to that of BiOIO3. This process can effectively suppress the recombination of electrons and holes. According to the active species trapping results, more ·OH are generated with visible-light irradiation than with UV light irradiation, which may be attributed to intense photoabsorption of AgI in visible region. Thus, remarkably enhanced photocatalytic performance is achieved for AgI/BiOIO3 heterostructure under visible light illumination.
In summary, AgI/BiOIO3 heterostructure was fabricated by a facile in-situ precipitation route. In comparison with pristine AgI and BiOIO3, AgI/BiOIO3 exhibited remarkably enhanced photocatalytic activity for degradation of MO under irradiation of both UV and visible light, which is due to the high charge separation derived from heterostructurefabrication. The different photoreactivity enhancement levels under UV and visible light were also demonstrated, which correspond to the different photocatalytic mechanisms. Importantly, AgI/BiOIO3 shows a universal photo-oxidation ability in decomposing 2, 4-DCP, phenol, bisphenol A, tetracycline hydrochloride and chlortetracycline hydrochloride under visible light, and a strong mineralization ability for decomposing BPA into CO2 and H2O was revealed. This study provides a promising heterostructurephotocatalyst for efficiently treating industrial contaminants and antibiotics.
AcknowledgmentsThis work was jointly supported by the National Natural Science Foundation of China (Nos. 51672258, 51572246, 51572270, U1662118), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB17030300), and the Fundamental Research Funds for the Central Universities (No. 2652015296).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.09.017.
| [1] |
M. Kapilashrami, Y.F. Zhang, Y.S. Liu, A. Hagfeldt, J.H. Guo, Chem. Rev. 114(2014) 9662-9707. DOI:10.1021/cr5000893 |
| [2] |
H.J. Yu, R. Shi, Y.X. Zhao, et al., Adv. Mater. 29(2017) 701-709. |
| [3] |
L. Shang, H. Yu, X. Huang, et al., Adv. Mater. 28(2015) 1668-1674. |
| [4] |
W.J. Wang, B.B. Huang, X.C. Ma, et al., Chem. Eur. J. 19(2013) 14777-14780. DOI:10.1002/chem.201302884 |
| [5] |
J.G. Yu, J.X. Low, W. Xiao, P. Zhou, M. Jaroniec, J. Am. Chem. Soc. 136(2014) 8839-8842. DOI:10.1021/ja5044787 |
| [6] |
C.T. Yip, H. Huang, L. Zhou, et al., Adv. Mater. 23(2011) 5624-5628. DOI:10.1002/adma.v23.47 |
| [7] |
A.B. Djurisic, X. Chen, Y.H. Leung, J. Mater. Chem. 22(2012) 6526-6535. DOI:10.1039/c2jm15548f |
| [8] |
H.W. Huang, K. Xiao, N. Tian, Y.X. Guo, Y.H. Zhang, RSC Adv. 5(2015) 81078-81086. DOI:10.1039/C5RA14405A |
| [9] |
H.F. Cheng, B.B. Huang, Y. Dai, Nanoscale 6(2014) 2009-2026. DOI:10.1039/c3nr05529a |
| [10] |
X. Zhang, Z.H. Ai, F.L. Jia, L.Z. Zhang, J. Phys. Chem. C 112(2008) 747-753. DOI:10.1021/jp077471t |
| [11] |
H.W. Huang, X. Han, X.W. Li, et al., ACS Appl. Mater. Interfaces 7(2015) 482-492. DOI:10.1021/am5065409 |
| [12] |
H.W. Huang, K. Xiao, Y. He, et al., App. Catal. B 199(2016) 75-86. DOI:10.1016/j.apcatb.2016.06.020 |
| [13] |
L.W. Zhang, Y. Man, Y.F. Zhu, ACS Catal. 1(2011) 841-848. DOI:10.1021/cs200155z |
| [14] |
A. Akhundi, A.H. Yang, RSC Adv. 6(2016) 106572-106583. DOI:10.1039/C6RA12414C |
| [15] |
H.W. Huang, Y. He, Z.S. Lin, L. Kang, Y.H. Zhang, J. Phys. Chem. C 117(2013) 22986-22994. DOI:10.1021/jp4084184 |
| [16] |
H.W. Huang, Y. He, X.W. Li, et al., J. Mater. Chem. A 3(2015) 24547-24556. DOI:10.1039/C5TA07655B |
| [17] |
H.W. Huang, S.B. Wang, Y.H. Zhang, X. Han, Mater. Res. Bull. 62(2015) 206-211. DOI:10.1016/j.materresbull.2014.11.032 |
| [18] |
Y. He, H.W. Huang, Y.H. Zhang, et al., Mater. Res. Bull. 64(2015) 405-409. DOI:10.1016/j.materresbull.2015.01.011 |
| [19] |
Y. He, H.W. Huang, X.W. Li, et al., Colloids Surf. A 467(2015) 195-200. DOI:10.1016/j.colsurfa.2014.11.007 |
| [20] |
S.B. Wang, H.W. Huang, Y.H. Zhang, Solid. State Sci. 62(2016) 43-49. DOI:10.1016/j.solidstatesciences.2016.10.015 |
| [21] |
X. Wang, J.C. Ran, M. Tao, et al., Mater. Sci. Semi. Process 41(2016) 317-322. DOI:10.1016/j.mssp.2015.09.021 |
| [22] |
Y. He, Y.H. Zhang, H.W. Huang, et al., Colloids Surf. A 462(2014) 131-136. DOI:10.1016/j.colsurfa.2014.07.034 |
| [23] |
H.W. Huang, Y. He, R. He, et al., Inorg. Chem. Commun. 40(2014) 215-219. DOI:10.1016/j.inoche.2013.12.024 |
| [24] |
S.D. Nguyen, J. Yeon, S.H. Kim, P.S. Halasyamani, J. Am. Chem. Soc. 133(2011) 12422-12425. DOI:10.1021/ja205456b |
| [25] |
C. Zeng, Y.M. Hu, Y.X. Guo, et al., ACS Sustain. Chem. Eng. 4(2016) 3305-3315. DOI:10.1021/acssuschemeng.6b00348 |
| [26] |
H.W. Huang, Y. He, Y.X. Guo, et al., Solid State Sci. 46(2015) 37-42. DOI:10.1016/j.solidstatesciences.2015.05.008 |
| [27] |
Y.C. Yang, J.W. Wen, J.H. Wei, et al., ACS Appl. Mater. Interfaces 5(2013) 6201-6207. DOI:10.1021/am401167y |
| [28] |
G.Q. Luo, X.J. Jiang, M.J. Li, et al., ACS Appl. Mater. Interfaces 5(2013) 2161-2168. DOI:10.1021/am303225n |
| [29] |
Y.S. Lee, H.J. Lee, W.S. Choi, Langmuir 30(2014) 9584-9590. DOI:10.1021/la502378z |
| [30] |
C. Zeng, Y.M. Hu, Y.X. Guo, et al., Appl. Catal. B 194(2016) 62-73. DOI:10.1016/j.apcatb.2016.04.036 |
| [31] |
B. Chen, Y. Deng, H. Tong, J. Ma, Superlattices Microst. 69(2014) 194-203. DOI:10.1016/j.spmi.2014.01.019 |
| [32] |
S.X. Yu, H.W. Huang, F. Dong, et al., ACS Appl. Mater. Interfaces 7(2015) 27925-27933. DOI:10.1021/acsami.5b09994 |
| [33] |
X.M. Qi, M.L. Gu, X.Y. Zhu, et al., Chem. Eng. J. 285(2016) 11-19. DOI:10.1016/j.cej.2015.09.055 |
| [34] |
J.W. Feng, H.W. Huang, S.X. Yu, F. Dong, Y.H. Zhang, Phys. Chem. Chem. Phys. 18(2016) 7851-7859. DOI:10.1039/C5CP06685A |
| [35] |
N. Tian, H.W. Huang, Y.H. Zhang, Y. He, Y.X. Guo, RSC Adv. 4(2014) 42716-42722. DOI:10.1039/C4RA05917D |
| [36] |
N. Tian, Y.H. Zhang, H.W. Huang, Y. He, Y.X. Guo, J. Phys. Chem. C 118(2014) 15640-15648. DOI:10.1021/jp500645p |
| [37] |
D.H. Cui, X.C. Song, Y.F. Zheng, RSC Adv. 6(2016) 71983-71988. DOI:10.1039/C6RA14486A |
| [38] |
C. Zeng, H.W. Huang, F. Dong, et al., Appl. Catal. B 200(2017) 620-632. DOI:10.1016/j.apcatb.2016.07.029 |
| [39] |
Y.Y. Bu, Z.Y. Chen, ACS Appl. Mater. Interfaces 6(2014) 17589-17598. DOI:10.1021/am503578s |
| [40] |
S. Chen, Y. Hu, S. Meng, X. Fu, Appl. Catal. B 150(2014) 564-573. |
| [41] |
J. Shao, L. Tong, S. Tang, Z. Guo, H. Zhang, ACS. Appl. Mater. Interfaces 7(2015) 5391-5398. DOI:10.1021/am508881k |
| [42] |
W.J. Wang, H.F. Cheng, B.B. Huang, et al., J. Colloid. Interface Sci. 442(2015) 97-102. DOI:10.1016/j.jcis.2014.11.061 |
| [43] |
H. Li, H. Yu, X. Quan, S. Chen, Y. Zhang, ACS Appl. Mater. Interfaces 8(2016) 2111-2119. DOI:10.1021/acsami.5b10613 |