 2017, Vol. 28
2017, Vol. 28 



b College of Science, Beijing University of Chemistry Technology, Beijing 100029, China
The combustion of the fossil fuel not only leads to the growing worldwide energy crisis, but also causes serious environmental pollution problem, such as the acid deposition, haze and the formation of PM2.5 [1-4]. Consequently, researches on alternative and clean energy resources have become an urgent challenge for scientists all over the world. There are many possible alternative energy resources could be used as the new energy candidates, such as the solar, wind, geothermal and hydrogen [5]. As a renewable, green, safe and viable energy resource, hydrogen (H2), which owns a high gravimetric energy density, has been widely investigated as the next generation energy carrier during the past few decades [6-11].
Till now, many technologies have been developed to produce H2 [5, 12-17]. Water splitting by PEC process has been studied as an environmental friendly and potential route for the transformation from the inexhaustible solar energy to the directly used and green H2 energy [5, 12, 18-24]. However, in a typical PEC water splitting process, the half reaction on the photoanode is the O2 evolution reaction, whose product O2 is not of significant value. It is more energy efficient if we can replace the O2 evolution reaction with some more useful oxidation reaction. We thus propose that the wastewater treatment could be integrated to the PEC water splitting, which may make the PEC water splitting economically feasible [5, 8, 9, 25-28]. The PEC wastewater treatment has been reported by many researchers [8, 29]. In this process, the organics dye in wastewater is oxidized at the anode, while the H2 is generated at the cathode. Thus, H2 generation can be well integrated into the PEC of wastewater treatment simultaneously.
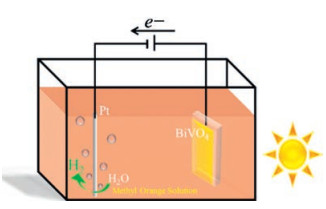
As an n-type semiconductor, bismuth vanadate (BiVO4), which owns a narrow band gap (2.4 eV) and a broad response in visible light range, has been considered as one of the most promising photoanode candidates for water splitting [18, 19, 30-32]. Besides, many references reported it has a high oxidation ability and excellent stability for oxidation degradation [33, 34]. Herein, we use a nanostructured BiVO4 film as the photoanode for the oxidation of azo dye with a simultaneously H2 production on the cathode. As shown in Scheme 1, a two electrodes configuration was employed with a Pt wire as the counter electrode, while BiVO4 as the working electrode, and electrolyte is the solution of azo dye in Na2SO4. During the PEC process, the oxidation of azo dye takes place on the anode, while water is reduced on the cathode to produce H2.

|
Download:
|
| Scheme 1. Experimental device for methyl orange degradation with simultaneous H2 generation via PEC route by nanostructured BiVO4. | |
{kind=link}
All chemical reagents are analytical grade and used without any further purification. Deionized water was used throughout the whole experiment. FTO glasses were sonicated by immersing in the acetone, ethanol and DI water in sequence to remove the impurities that existed in the surface of the FTO glasses.
The BiVO4/FTO was synthesized by a drop-coating method of the BiVO4 precursor solution onto the conducting plane of the FTO. In a typical procedure, 0.5 mmol of Bi(NO3)3·5H2O, 0.5 mmol NH4VO3 were dissolved in the mixture of 5 mL nitric acid and 5 mL DI water. After the solute was dissolved, 5 mL of ethanol was added to dilute the mixture. Then the BiVO4 precursor (40 μL) was dripped to the surface of FTO. After dried at 60 ℃ in an oven, the catalysts were calcined at 500 ℃ for 3 h with a ramping rate of 2 ℃/min.
The microstructure of as-prepared electrode was characterized with scanning electron microscopy (SEM, Philips XL30FEG, Netherlands). The crystal structure of the as-prepared electrode was measured using X-ray diffraction (XRD) patterns by a Druker with Cu Kα radiation ranging from 10° to 70°. UV–vis spectrum of the as-prepared electrode was performed using a UV–vis spectrophotometer (SHIMADZU UV-2600) with an integrating sphere attachment. The change in the total organic carbon (TOC) of MO solution after degradation was tested using TOC analyzer (TOC 2500, Shimazu, Japan). X-ray photoelectron spectroscopy (XPS) was performed on a Perkin-Elmer PHI-5000C ESCA system equipped with a dual X-ray source, using an Mg Kα radiation source and a hemispherical energy analyzer. Linear Sweep Voltammetry (LSV) measurements were carried out by an electrochemical station (CHI 660E, Shanghai Chenhua Co., Ltd. China).
During the photoelectrochemical tests, a standard three electrodes configuration were employed, the synthesized BiVO4 film is used as the working electrode, Pt wire as the counter electrode and Ag/AgCl as the reference electrode. The potential conversion from vs. Ag/AgCl to vs. reversible hydrogen electrode (RHE) was calculated using Eq. (1):

|
(1) |
The PEC degradation of organic contaminant and the amounts of H2 were performed in a PEC reactor in a two-electrode system at 3.5 V under an air mass (AM) 1.5 G radiation in a 200 mL reactor with 120 mL solution filled in. As the protective gas, Ar was bubbled into the reactor cell. The size of the synthesized mesoporous film electrode is 1 ×1 cm2. 300 W Xe lamp was used as the artificial solar light source and illuminate from the back side of the film. The working electrode was illuminated from the light source through a quartz window of the reactor cell. The bias voltage applied to the film electrode was supplied by the electrochemical a potentiostat. In this system, methyl orange (MO, 10 ppm) was used as an azo dye during the PEC process. And the Na2SO4 (0.5 mol/L) was used as the supporting electrolyte. The adsorption spectra and contents of the MO in the solution were measured by UV–vis spectrophotometer (SHIMADZU UV-2600). The degradation ration of MO was defined in Eq. (2):

|
(2) |
where C0 is the initial concentration of MO in the solution; Ct denotes the concentration of MO at the different moment. HPLC was performed by an Agilent 1100 Series. Samples were separated on a Zorbax SB C18 (4.6 mm×150 mm, 5 μm) reverse phase column at 50 ℃. The mobile phase consisting of methanol/water = 50:50 (v/v) was set at a flow rate of 0.8 mL/min. The concentration of H2 was analyzed by gas chromatography (SHIMADZU, GC-2014) using a thermal conductivity detector (TCD). The theoretical evolution rate of H2 is calculated according to the following equation (Eq. (3)):

|
(3) |
Where, v indicates the evolution rate of H2 (mol cm-2 h-1); I indicates the average current (A); t indicates the time (s); Z indicates the transferred electron number (1); F is the Faraday's constant (96500 C/mol); A is the area of the BiVO4 film (cm2).
The equation for the calculation of Faradaic efficiency is described in Eq. (4):

|
(4) |
Where, m is the experimental value of H2 (mol); n is the reacted electron number (1); F is the Faraday's constant (96500 C/mol); I is the average current (A); t is the time (s).
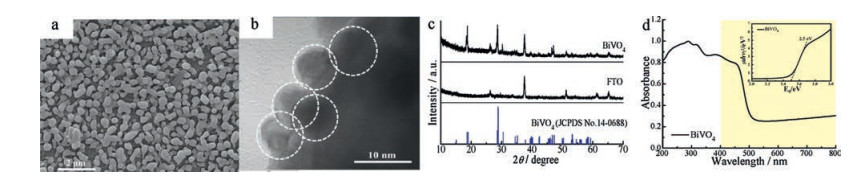
The microstructure of the as-prepared BiVO4 film is investigated by SEM in Fig. 1a. The film of BiVO4 is composed of spherical naoparticles with diameter of about 500 nm. And the sphere consisted of nanoparticles, with diameter of about 9 nm (Fig. 1b). A BiVO4 nanofilm can be successfully synthesized by this simple drip-coating method, the nanofilm shows porous structure, ultrathin thickness and high surface area, which are beneficial for the photoelectrocatalytic oxidation reaction. The XRD experiment is carried out to determine the chemical composition and crystal structure of the photoelectrode, and the obtained XRD spectra of BiVO4 and naked FTO are shown in Fig. 1c. It is clear that the diffraction patterns of the synthesized BiVO4 is highly consistent with the characteristic reflections of the monoclinic scheelite structure (JCPDS No. 14-0688), and the sharp peaks of BiVO4 film sample indicate it has a good crystallinity. The thickness of the as-prepared BiVO4 film is thin, as a result, the diffraction peaks of FTO substrate are also observed.

|
Download:
|
| Fig. 1. (a) SEM images (b) HRTEM image of nanostructured BiVO4 film; (c) XRD patterns of BiVO4 and naked FTO; (d) UV–vis absorption spectrum and the Tauc plot of BiVO4 (insert). | |
{kind=link}
UV-vis diffraction reflection spectroscopy (UV-vis DRS) is conducted to investigate the optical property of the synthesized BiVO4 film. As shown in Fig. 1d, three broad absorption bands of the synthesized BiVO4 film are observed at wavelengths of 200– 350 nm, 350–420 nm, and 420–500 nm, respectively. Besides, there is a strong absorption in the visible light region (λ > 420 nm) for the synthesized BiVO4 film. And, the band gap of the BiVO4 film calculated from the Tauc plot is 2.5 eV (Fig. 1d inset), which is in consistence with the reported band gap of BiVO4 [18]. The BiVO4 film with an intense absorption from the UV to visible light range could be favor for the efficient utilization of the solar light.
The PEC performances are evaluated both in dark and under AM 1.5 G simulated solar light illumination (100 mW/cm2) in the MO solution. Fig. S1a in Supporting information displays the photocurrent density–bias (J-V) curves of BiVO4 electrode film in dark and under irradiation by using a three-electrode configuration. It is clearly that the current density is negligible in the dark condition. The detected current increased remarkably when light is turnedon. The nanostructured BiVO4 electrode film produced an anodic photocurrent demonstrating the n-type nature of BiVO4 [35]. The transient photocurrent performance is performed to preliminary estimate the charge carrier recombination properties on the interface of the semiconductor. As shown in Fig. S1b, the photocurrent value presents fast and uniform results, alternatively increasing to maximum value and decreased to zero corresponding to the light switched on and off. Besides, the phenomenon of the photoresponse is reversible. It is known that the generated transient anodic photocurrent wave, when the light is turn-on, represents the accumulation of holes at the electrode-electrolyte interface. At the same time, the generated transient cathodic photocurrent wave, when the chopped light is turn-off, represents the recombination of electrons from the conduction band with the accumulated holes. The obtained nanostructured BiVO4 displays an outstanding photocurrent density compared with the published researches [36, 37].
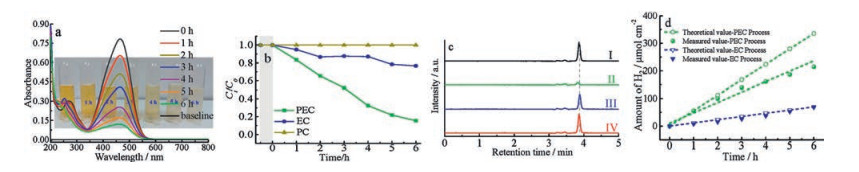
The catalytic activities of the typical azo dye samples are investigated using the degradation of MO at room temperature. The PEC (at a bias potential of 3.5 V with irradiation), EC (electrocatalytic process at a bias potential of 3.5 V, without irradiation), and PC (photocatalytic, without a bias potential) in neutral aqueous solutions are performed under the given conditions. The UV–vis absorption spectra of MO solution are conducted under wavelengths from 200–800 nm during the PEC, EC and PC operations (Fig. 2a and Fig. S2a-b in Supporting information). The MO solution was stirred for 30 min to reach the adsorption equilibrium. The changes of MO concentration versus the reaction time over BiVO4 film during the PEC, EC and PC process were shown in Fig. 2a and Figs. S2a-b in Supporting information, respectively. The major characteristic peak of MO (around 464 nm) diminishes gradually under the PEC and EC process. However, the PC process test shows that MO is stable under visible light irradiation, indicating that the photolysis of MO is negligible during the whole catalytic period. Meanwhile, the color of the suspension changed gradually during the PEC and EC process, suggesting that the chromophoric structure of MO was decomposed. Additionally, new absorbance peaks that appeared ranging from 210–250 nm indicated the formation of small molecular intermediate products in Fig. 2a. Hence, the activity order of MO degradation is follows the tendency of PEC > EC > PC. The relative concentration (Ct/C0, Ct and C0 stand for the remnant and initial concentrations of MO, respectively) as a function of time is shown in Fig. 2b. As shown in Fig. 2b, the BiVO4 film exhibits the highest catalytic activity via PEC process, which significantly outperformed the same film under the EC and PC process. To further investigate the degradation kinetics, the pseudo-first-order kinetic model (ln (Ct/C0) = kt, k is the kinetic constant, t is the reaction time) is applied to fit the degradation data and the fitting results are shown in Table S1 in Supporting information. As seen in Fig. S2c and Table S1, for the degradation rate of MO, the PEC process obtains the highest rate constant of about 0.3207 h-1, which is higher than that of EC (about 0.0421 h-1) or PC (about 0.005 h-1). The results indicate that the degradation efficiency of MO can be significantly improved under the combination of the photocatalytic process and the electrocatalytic process, which could effectively facilitate the separation of photogenerated electron-hole pairs.

|
Download:
|
| Fig. 2. (a) The temporal evolution of UV-vis adsorption spectra and the corresponding optical images of MO solutions at different irradiation time by PEC degradation using the as-prepared BiVO4 electrode film during visible light irradiation; (b) comparison of degradation performance by PEC, EC and PC process; (c) HPLC chromatograms of the initial MO solution (Ⅰ) and after 6 h degradation over PEC (Ⅱ), EC (Ⅲ) and PC (Ⅳ) routes; (d) Amount of hydrogen at an applied potential of 3.5 V in two-electrode system for 6 h. | |
{kind=link}
HPLC chromatograms of the MO solution at initial, and after different catalytic route for 6 h are recorded in Fig. 2c. It is remarkably to note that a much lower intensity of MO is presented by PEC route than that of EC or PC route. Besides, the retention time of MO moves forward, indicating the existence of obtained small molecule intermediate from MO during the PEC process. The HPLC results further confirm that the BiVO4 electrode film exhibits much more excellent degradation performance of MO degradation by PEC process as compared with EC or PC process. Despite its effectiveness in MO decoloration, the catalytic degradation on the BiVO4 film may not lead to the mineralization. The mineralization depends on whether the refractory intermediate compounds are more difficult to degrade relative to the original substrate. Therefore, TOC measurements are conducted under different catalytic degradation process. As shown in Fig. S2d, 66% of the TOC is removed after discoloration of MO for 6 h with degradation by PEC process. However, with PC process the mineralization of MO took place at a much lower rate and TOC was reduced nearly 5%. Obviously, the rate of the decolorization was much faster than that of mineralization.
The H2 evolution is also studied at the same time with the MO degradation during the PEC and EC process. Fig. 2d shows the H2 amount evolved theoretically (calculated from the photocurrent) vs. evolved experimentally by PEC or EC process, and the current density during these two processed are provided as well. In addition, the current densities, theoretical evolution rates of H2, experimental evolution rates of H2 and the Faradaic efficiencies of H2 during the PEC and EC process are summarized in Table S2 in Supporting information. As shown in Fig. 2d and Table S2 in Supporting information, the theoretical value and the measured value during the PEC process is much higher than that of the EC process. The H2 evolution rate by PEC process is 34.44 μmol h-1 cm-2, which is 3 times more efficient than the EC process (11.37 μmol h-1 cm-2). However, some of the generated electrons on the cathode can be consumed by the reduction of the small intermediates formed by the degradation of MO, consequently, an obvious difference between the theoretical H2 evolution rate and measured H2 evolution rate during the PEC process is observed. The calculated Faradic efficiency of H2 during the PEC process (62%) is thus lower than that of EC process (96%).
Besides, the stability of the H2 generation is confirmed by the curves of photocurrent density vs. time, which is steady and shows ignorable changes in 6 h H2 experiment with BiVO4 as the anode material during the PEC and EC processes (Fig. S3 in Supporting information). The stability is further confirmed by performing the MO degradation for five runs by PEC route. These experiments are carried out by applying the recycled BiVO4 film to fresh MO solutions with the same concentration, and the results are shown in Fig. S4 in Supporting information. Since there is no obvious decrease in activity is observed after five runs, indicating that the BiVO4 film owns outstanding stability and recyclability during the PEC reaction and it would be a promising candidate for the wastewater treatment and H2 production. Additionally, the nanostructured BiVO4 photocatalyst shows more excellent recycle performance than previous reports [33, 38].
To investigate the origin of the enhanced PEC activity, the electrochemical property of BiVO4 is performed by the electrochemical impedance spectroscopy (EIS). The EIS measurements are conducted at the same electrolyte solution. (Fig. S5a in Supporting information) shows the EIS Nyquist plots of BiVO4 film in the dark and under the light illumination. Here, the EIS results are obtained by using a three-electrode cell system, thus the arc radius in Nyquist plot represents the charge transfer resistance on the working electrode interface. The diameter of the arc radius on the Nyquist plot in the presence of solar light (PEC process) is smaller than that in the absence of solar light (EC process), indicating the efficient charge separation and transport. Hence, the nanostructured BiVO4 electrode film exhibited outstanding catalytic performance of degradation of azo dye and H2 generation during the PEC process than that of EC process. Besides, the Bode phase plot of nanostructured BiVO4 is shown in Fig. S5b. The result indicates that there is no current passing through the external circuit. And the nanostructured BiVO4 shows the highest Bode phase at 0.25 Hz.
In order to detect the micromorphology, structure and surface of the catalysis after PEC degradation process, SEM image, XRD pattern and XPS analysis of the photoanode were conducted after PEC degradation process. As shown in Fig. S6a and b in Supporting information, the micromorphology and structure keep unchanged after PEC degradation process. Besides, after comparing the surface elements of the photocatalyst before and after catalytic action with XPS measurement (Fig. S6c), hardly any new elements could be detected or any element disappeared. The above results indicate that the physicochemical property of the nanostructured BiVO4 could not be destroyed during the PEC degradation process.
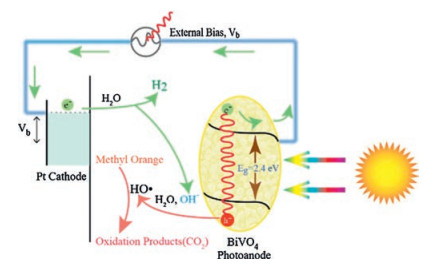
This PEC system involving a photocatalytic oxidation process on the anode and a reduction process on the cathode is shown in Scheme 2. The half reactions on the photoanode side are described as follows: with the xenon light irradiation, the electrons of BiVO4 film are excited from the valence band (VB) to the conduction band (CB), which producing positive holes (h+) in the VB and negative electrons (e-) in the CB (Eq. (5)). To avoid the recombination of photogenerated h+ and e- and provide enough energy potential for H2 generation, an external bias is introduced. Then the photogenerated e- travels through the interface of BiVO4 electrode to the cathode by external circuit, while h+ is involved in the oxidation reaction of the MO (Eq. (8)), via ·OH radicals formed through the reactionwith OH- and H2O in the solution (Eqs. (6) and (7)). For the MO degradation, the ·OH radicals attack the azo band in the MO molecule and break down the N-N bond, and then the intermediates are degraded to small molecules and finally oxidized to CO2 and H2O [39]. On the cathode of the PEC system, H2O as terminal electron acceptors is reduced to H2 and OH- by accepting the electrons from cathode with assistance of Pt wire (Eq. (9)). OH- is then generated on the cathode for the continuously consuming on the photoanode side, forming the PEC process circle.

|
(5) |

|
(6) |

|
(7) |

|
(8) |

|
(9) |

|
Download:
|
| Scheme 2. Schematic illustration of MO degradation with simultaneous H2 generation via PEC route. | |
{kind=link}
In summary, we prepared a nanostructured BiVO4 film electrode for the degradation of azo dye and simultaneously H2 production by PEC process. The BiVO4 electrode presented a strong absorption from the UV to visible light range. The PEC process shows excellent catalytic activity of MO degradation and also H2 production, which are both much higher than EC or PC process alone. We show that the wastewater treatment can be well integrated into the PEC water splitting, which may make the PEC water splitting economically feasible. The proposed PEC system shows the potential to be a sustainable and energy-saving way for simultaneous wastewater treatment and energy recovery.
AcknowledgmentsThe authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos. 21507011, 21677037, 21607027), China Postdoctoral Science Foundation (No. KLH1829017), and Ministry of Science and Technology of China (Nos. 2016YFE0112200, 2016YFC0203700).
| [1] |
S. Hameed, R.D. Cess, J.S. Hogan, J. Geophys. Res. Oceans 85(1980) 7537-7545. DOI:10.1029/JC085iC12p07537 |
| [2] |
J.J. Griffin, E.D. Goldberg, Geochimica. ET. Cosmochimica. Acta 45(1981) 763-769. DOI:10.1016/0016-7037(81)90047-8 |
| [3] |
H. Hevy, W.J. Moxim, Tellus. B 41(1989) 256-271. DOI:10.3402/tellusb.v41i3.15078 |
| [4] |
S.S. Penner, Energy 16(1991) 1417-1419. DOI:10.1016/0360-5442(91)90010-J |
| [5] |
J.Y. Jiang, M. Chang, P. Pan, Environ. Sci. Technol. 42(2008) 3059-3063. DOI:10.1021/es702466k |
| [6] |
C.A. Reddy, M.P. Bryant, M.J. Wolin, J. Bacteriol. 110(1972) 126-132. |
| [7] |
X. Zong, H.J. Yan, G.P. Wu, et al., J. Am. Chem. Soc. 130(2008) 7176-7177. DOI:10.1021/ja8007825 |
| [8] |
B. Cao, G.S. Li, H.X. Li, Appl. Catal. B:Environ. 194(2016) 42-49. DOI:10.1016/j.apcatb.2016.04.033 |
| [9] |
Z.W. Chen, X.Y. Jiang, C.B. Zhu, C.K. Shi, Appl. Catal. B:Environ. 199(2016) 241-251. DOI:10.1016/j.apcatb.2016.06.036 |
| [10] |
E. Gianotti, M. Taillades-Jacquin, Á. Reyes-Carmona, et al., Appl. Catal. B:Environ. 185(2016) 233-241. DOI:10.1016/j.apcatb.2015.12.015 |
| [11] |
D.S. Hu, Y. Xie, L.J. Liu, et al., Appl. Catal. B:Environ. 188(2016) 207-216. DOI:10.1016/j.apcatb.2016.01.069 |
| [12] |
B.H. Wu, D.Y. Liu, S. Mubeen, et al., J. Am. Chem. Soc. 138(2016) 1114-1117. DOI:10.1021/jacs.5b11341 |
| [13] |
H.H. Liu, D.L. Chen, Z.Q. Wang, H.J. Jing, R. Zhang, Appl. Catal. B:Environ. 203(2017) 300-313. DOI:10.1016/j.apcatb.2016.10.014 |
| [14] |
Y.F. Zhao, B. Zhao, J. Liu, et al., Angew. Chem. Int. Ed. 55(2016) 4215-4219. DOI:10.1002/anie.201511334 |
| [15] |
Y.F. Zhao, X.D. Jia, G.I.N. Waterhouse, et al., Adv. Energy Mater 6(2016) 1501974. DOI:10.1002/aenm.201501974 |
| [16] |
L. Shang, B. Tong, H.J. Yu, et al., Adv. Energy Mater. 6(2016) 1501241. DOI:10.1002/aenm.201501241 |
| [17] |
J.M. Liu, L. Han, H.Y. Ma, et al., Sci. Bull. 61(2016) 1543-1550. DOI:10.1007/s11434-016-1162-3 |
| [18] |
L.W. Zhang, C.Y. Lin, V.K. Valev, et al., Small 10(2014) 3970-3978. DOI:10.1002/smll.201400970 |
| [19] |
S.M. Sun, W.Z. Wang, D.Z. Li, L. Zhang, D. Jiang, ACS Catal. 4(2014) 3498-3503. DOI:10.1021/cs501076a |
| [20] |
J. Seo, T. Takata, M. Nakabayashi, et al., J. Am. Chem. Soc. 137(2015) 12780-12783. DOI:10.1021/jacs.5b08329 |
| [21] |
T. Reier, Z. Pawolek, S. Cherevko, et al., J. Am. Chem. Soc. 137(2015) 13031-13040. DOI:10.1021/jacs.5b07788 |
| [22] |
F. Jiang, T. Gunawan Harada, et al., J. Am. Chem. Soc. 137(2015) 13691-13697. DOI:10.1021/jacs.5b09015 |
| [23] |
L.L. Feng, G.T. Yu, Y.Y. Wu, et al., J. Am. Chem. Soc. 137(2015) 14023-14026. DOI:10.1021/jacs.5b08186 |
| [24] |
C.A. Downes, S.C. Marinescu, J. Am. Chem. Soc. 137(2015) 13740-13743. DOI:10.1021/jacs.5b07020 |
| [25] |
P.A. Williams, C.P. Ireland, P.J. King, et al., J. Mater. Chem. 22(2012) 20203-20209. DOI:10.1039/c2jm33446a |
| [26] |
L. Wang, M.H. Cao, Z.H. Ai, L.Z. Zhang, Environ. Sci. Technol. 48(2014) 3354-3362. DOI:10.1021/es404741x |
| [27] |
K.X. Li, Z.X. Zeng, L.S. Yan, et al., Appl. Catal. B:Environ. 187(2016) 269-280. DOI:10.1016/j.apcatb.2016.01.046 |
| [28] |
J. Ke, J. Liu, H.Q. Sun, et al., Appl. Catal. B:Environ. 200(2017) 47-55. DOI:10.1016/j.apcatb.2016.06.071 |
| [29] |
M. Panizza, P.A. Michaud, G. Cerisola, C. Comninellis, Electrochem. Commun. 3(2001) 336-339. DOI:10.1016/S1388-2481(01)00166-7 |
| [30] |
H. Tong, S.X. Ouyang, Y.P. Bi, et al., Adv. Mater. 24(2012) 229-251. DOI:10.1002/adma.201102752 |
| [31] |
K.J. McDonald, K.S. Choi, Energy Environ. Sci. 5(2012) 8553-8557. DOI:10.1039/c2ee22608a |
| [32] |
M. Zhou, J. Bao, Y. Xu, et al., ACS Nano 8(2014) 7088-7098. DOI:10.1021/nn501996a |
| [33] |
J.L. Zhang, Y. Lu, L. Ge, et al., Appl. Catal. B:Environ. 204(2017) 385-393. DOI:10.1016/j.apcatb.2016.11.057 |
| [34] |
J. Bai, R. Wang, Y.P. Li, et al., J. Hazard. Mater. 311(2016) 51-62. DOI:10.1016/j.jhazmat.2016.02.052 |
| [35] |
S.K. Pilli, T.E. Furtak, L.D. Brown, et al., Energy Environ. Sci. 4(2011) 5028-5034. DOI:10.1039/c1ee02444b |
| [36] |
J.Q. Li, J. Zhou, H.J. Hao, L.W. Jie, Appl. Surf. Sci. 399(2017) 1-9. DOI:10.1016/j.apsusc.2016.12.048 |
| [37] |
N. Myung, W. Lee, C. Lee, S. Jeong, K. Rajeshwar, ChemPhysChem 15(2014) 2052-2057. DOI:10.1002/cphc.v15.10 |
| [38] |
E. Aguilera-Ruiz, U.M. Garcia-Perez, M. Garza-Galvan, et al., Appl. Surf. Sci. 328(2015) 361-367. DOI:10.1016/j.apsusc.2014.12.059 |
| [39] |
S.B. Xu, Y.F. Zhu, L. Jiang, Y. Dan, Water Air Soil Pollut. 213(2010) 151-159. DOI:10.1007/s11270-010-0374-4 |