 2017, Vol. 28
2017, Vol. 28 



The goal of pharmaceutical research and development is to discover and develop drug products with safety, efficacy and costeffectiveness. Besides the active pharmaceutical ingredient (API), multiple types of inactive ingredients (i.e. excipients) were also incorporated into the final drug products to optimize the drug delivery profile. Therefore, the final drug product is always a multicomponent and usually multi-phase material system. The interdependent relationship between its formulation, processing, structure, property and performance places pharmaceutical research and development into the realm of materials science [1].
For those drugs to be delivered as solids, the selection of their specific solid forms plays a key role in controlling their physicochemical properties, such as solubility, dissolution rate, thermal stability, mechanical properties, and consequently bioavailability and processibility [1-4]. The solid forms available for drugs can be classified according to their composition (single-/ multi-component) and crystalline state (crystalline/amorphous) (Table 1). The drug itself can exist in the crystalline or amorphous state. The former sometimes exhibits polymorphic forms with different molecular packing [5-8], and the latter is generally an organic molecular glass [9-12]. Compared with the singlecomponent solid forms, incorporation of the second components into drug solid forms provides more structural variables and hence higher adjustability to their properties and performance. The conventionally used coformers for multi-component drug solid forms are small molecules. By cocrystallization with neutral molecules, co-crystals [13-16] or solvates/hydrates [17-19] of drugs can be obtained. If proton transfer occurs between the drug molecule and the coformer, ionized species will exist in the crystal lattice and therefore form a pharmaceutical salt [20-23]. Generally, the incorporation of small molecular coformers produces crystalline drug solids, but co-amorphous systems and amorphous salts are also possible [24-27].
|
|
Table 1 Classification of drug solid forms based on their composition and crystalline states. |
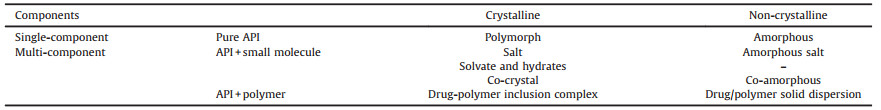
In addition to the small molecules, polymers represent another type of coformers in drug solids (Table 1). For example, drug molecules can be uniformly dispersed into the polymer matrix, where the drug crystallization may be kinetically inhibited, generating amorphous solid dispersions (Fig. 1A) [28-31]. In contrast, recent studies have found that polymers can also cocrystallize with drugs: linear polymers with appropriate chain size can be incorporated into the channel lattices constructed by drug molecules, forming drug-polymer inclusion complexes (IC, Fig. 1D), which is the focus of the current review [32-34].

|
Download:
|
| Fig. 1. Possible structures of a drug/polymer 2-component system. The red hexagons and blue curves represent drug molecules and polymer chains, respectively. The polymer chains can exist in amorphous state (random coils) or form crystalline lamella (folded lines). It should be noted that only crystalline polymers (e.g., poly(ethylene glycol) (PEG) and poly(ε-caprolactone) (PCL)) have the possibility of forming the two-phase structures shown in E and F. | |
{kind=link}
Compared with the conventional drug solid forms comprising pure API or API with small molecular coformers, the incorporation of long-chain polymers produces drug solid forms with distinctly different structures. In principle, a drug/polymer 2-component system can form totally 6 different structures (Fig. 1). When the drug is molecularly dispersed within the polymer matrix, an amorphous solid dispersion is obtained (Fig. 1A). For most drugpolymer pairs, the equilibrium solubility of drugs in the polymer matrix is generally very low. Therefore, the amorphous solid dispersion can be regarded as a supersaturated solution, and the drug molecules tend to precipitate out during manufacturing and storage. This can result in several different two-phase structures, with the drug phase or polymer phase being crystalline or amorphous (Figs. 1B, C, E and F).
In contrast to the aforementioned 2-phase structures, the drugpolymer ICs are actually in one homogeneous, crystalline phase, in which the drug molecules form a crystal lattice framework with parallel and isolated channels, and polymer chains of extended conformations reside in such channels. As a new pharmaceutical solid form, the generality and structural features of drug-polymer ICs are yet to be clearly understood, and their structure-property relationships, as well as potential applications deserve more studies.
2. Drug-polymer crystalline inclusion complexesAlthough so far only a few drug-polymer ICs have been reported in the pharmaceutical field (Fig. 2), crystalline ICs formed between small molecular hosts and polymer guests have been widely studied in the polymer science, among which urea and cyclodextrin systems are well-known examples [35-38].

|
Download:
|
| Fig. 2. Molecule structures of mavacoxib, griseofulvin, diflunisal, nevirapine, carbamazepine, poly(ethylene glycol) (PEG), poly(ε-caprolactone) (PCL) and poly (butylene adipate) (PBA). | |
{kind=link}
In the pharmaceutical field, the anti-inflammatory drug mavacoxib was reported in a patent application (published in 2006) to form crystalline complexes with polymers (i.e., oligo (ethylene glycol)s), which seemed to be the first drug-polymer IC in the literature [39]. However, similar structure was reported earlier for the resorcinol-poly(ethylene glycol) (PEG) complex [40, 41], though the pharmaceutical activity of resorcinol was never mentioned in these publications. In the years of 2011-2013 when we were studying the drug crystallization behavior in the polymer melt, we accidently observed that an antifungal drug griseofulvin was able to form crystalline ICs with PEG [32]. The formation of drug-polymer ICs was further confirmed in other drug systems including diflunisal [33], nevirapine and carbamazepine. And besides PEG, other linear polymers such as poly(ε-caprolactone) (PCL), poly(tetrahydrofuran) (PTHF), and poly(butylene adipate) (PBA) were also found to be incorporated into the channel lattices constructed by these drug molecules (Fig. 2).
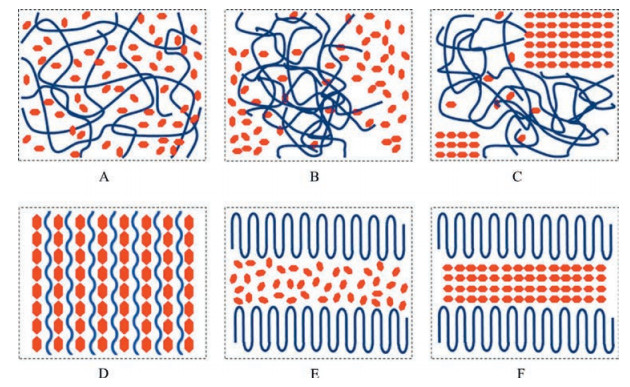
Similar to the widely studied ICs formed between small molecular hosts and polymer guests, in a drug-polymer IC, the drug molecules form a crystal lattice with parallel and isolated channels, which can accommodate polymer chains with extended conformations (Fig. 3, re-drawn based on the cif documents of Ref. [33], [46] and [47]). In contrast to the specific interactions (e.g., hydrogen bonds) commonly observed between drug molecules and small molecular coformers in co-crystals [42-44], in the drugpolymer ICs studied so far, it seems that van der Waals interactions between drug molecules and polymer chains are the dominant interactions to stabilize the otherwise possibly unstable IC channel framework. Therefore, the polymer chains in ICs mainly act as void fillers, similar to the solvent molecules in non-stoichiometric solvates [45]. Consequently, the same channel structure may accommodate different polymer guests with suitable chain crosssection sizes, thus forming isostructural ICs. It should be noted that for these drug-polymer ICs studied so far, only polymer chains without side groups were found to fit in the channels, which may be due to the limited cross-section sizes of the channel voids. However, at least in principle, linear polymers with side groups (e.g., polylactide) may also be incorporated into IC channels if the drug molecules can form a channel structure with sufficiently large internal diameter, which is one of the focuses in the future studies.

|
Download:
|
| Fig. 3. Channel frameworks formed by drug molecules in (A) diflunisal-polytetrahydrofuran IC, (B) nevirapine hemiethyl acetate solvate and (C) carbamazepine Form Ⅱ. | |
{kind=link}
As a new pharmaceutical solid form, drug-polymer ICs have been demonstrated for multiple drugs, however, their generality is not clear yet. Therefore, development of an effective search strategy for potential drug candidates of drug-polymer ICs would be useful. From the above studies on drug-polymer ICs, it was found that the drug-polymer ICs possess almost the same channel structures with their isostructural channel-type solvates. Similar phenomena were also observed for other extensively studied IC systems, such as urea [48], perhydrotriphenylene (PHTP) [49], and tris(o-phenylenedioxy) cyclotriphosphazene (TPP) [50, 51]. The close structural similarities between small molecule-polymer ICs and their corresponding solvates indicate that the guest molecules, both polymers and solvents, mainly act as void fillers to stabilize the host framework. The host channels originally filled by solvent molecules can also be occupied by polymer chains with suitable cross-section sizes. Therefore, the formation of isostructural channel-type solvates, which is widely reported in the crystal databases, may help to find drug candidates with intrinsic IC nature (Fig. 4). It is interesting to note that in the presence of both suitable guest polymers and solvents, drugs usually prefer to form inclusion complexes with polymers, probably because of the multivalent effect, which in turn is due to the smaller penalty in translational entropy of the polymers compared with small molecular solvents during the complex formation.

|
Download:
|
| Fig. 4. Schematic diagram showing the search strategy for drug-polymer ICs. Adapted with permission [33]. Copyright 2016, American Chemical Society. | |
{kind=link}
The drug-polymer ICs can be prepared through the same methods commonly used for pharmaceutical co-crystals, e.g., by cocrystallization of the two components in melt [32], in solution [33], or in a spray drying process. As for the structure confirmation, due to the isostructural relationship, drug-polymer ICs exhibit Xray diffraction patterns similar to those of the channel-type solvates. Single crystal X-ray diffraction provides direct evidence for the IC formation. However, for drug-polymer ICs, the exact conformation of the guest polymer chains cannot be solved so far due to their disordered state. Alternatively, solid-state NMR seems a powerful tool to investigate the structural features of the IC crystals. For example, the inclusion of guest polymers into host channels can be revealed by the 1H-13C host-guest correlation signals in their 2D 1H-13C HETCOR NMR spectrum [52], or by the averaged relaxation times of the host and guest in their 1H T1 and T1ρ relaxation measurements [32].
3. Structure-property relationshipsIn drug-polymer ICs, the guest polymers mainly act as void fillers. Different linear polymers may fit in the same channel structure and form isostructural ICs with the same drug. Therefore, the physicochemical properties and consequently the bioavailability of drugs may be optimized by simply changing the type and molecular weight (MW) of the guest polymers without substantially altering the crystal structure.
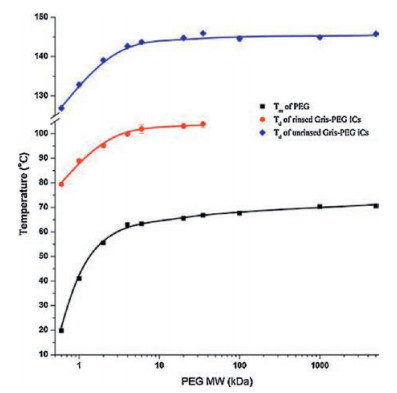
3.1. Thermal stabilityFor the commonly used co-crystals, their melting points, or dissociation temperatures, are usually in between, or sometimes lower than those of the drug and the coformer [13]. Similar to those co-crystals, drug-polymer ICs generally exhibit dissociation temperatures in between the melting points of their components, and interestingly, the dissociation temperature was further found to depend on the MW of the incorporated guest polymers.
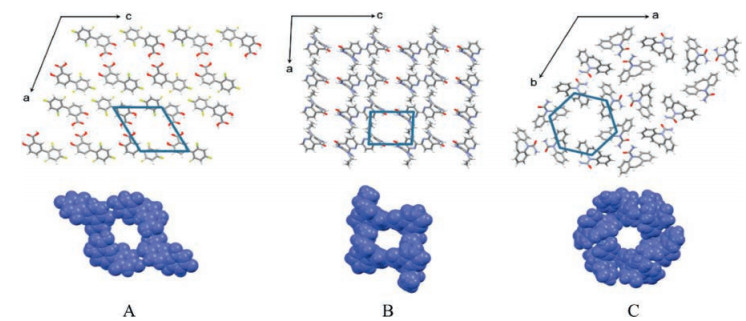
For example, griseofulvin-PEG ICs showed a phase transition to griseofulvin Form I during heating, which was associated with the dissociation of PEG and simultaneous recrystallization of griseofulvin [32, 34]. The dissociation temperatures of griseofulvin-PEG ICs first increased with PEG MW and finally reached a plateau when PEG MW reached 6 kDa (Fig. 5). This trend can be explained by the fact that the ends of PEG chains create crystal defects in the IC channels, and ICs with higher PEG MW will have lower chainend defect concentration and hence higher thermal stability [34]. Similar phenomenon was also observed for diflunisal-polymer ICs, with the dissociation temperature increasing with the chain length of guest polymers [33].

|
Download:
|
| Fig. 5. The variation of dissociation and melting temperatures of griseofulvin-PEG ICs with PEG MW. Adapted with permission [34]. Copyright 2016, Elsevier.. | |
{kind=link}
In addition to the polymer MW, different types of guest polymers are likely to produce different drug-polymer interactions and thus different thermal stability of the resulting IC crystals. Griseofulvin was found to form isostructural ICs with linear guest polymers PEG and PCL. Though both PEG and PCL themselves exhibited similar melting points, their corresponding griseofulvin IC crystals had distinctly different dissociation temperatures. For example, when the MW of the polymer were 6 kDa, the dissociation temperature of the griseofulvin IC formed with PCL is about 40 ℃ higher than that formed with PEG. The variation of the thermal stability of griseofulvin ICs with different guest polymers may arise from the different strength of drug-polymer interactions in the host channels.
3.2. Dissolution profilesDissolution behavior, including the dissolution rate and solubility, is another important issue that needs to be carefully considered for a pharmaceutical solid form. For a multi-component crystal (e.g., pharmaceutical co-crystals), the incorporation of a hydrophilic coformer can actually improve the solubility and dissolution rate of poorly water-soluble drugs [3, 53-55], while the use of hydrophobic coformers may lower the solubility and dissolution rate of drugs [53, 56]. Similar to the hydrophilic or hydrophobic coformer effects, the dissolution behavior of drugpolymer ICs can also be effectively modified by using hydrophilic or hydrophobic polymers as the guest.
Through the incorporation of water-soluble PEG, griseofulvin ICs showed faster dissolution and higher apparent aqueous solubility than both amorphous and crystalline griseofulvin [34]. In contrast to the aforementioned MW dependence of the thermal stability, the dissolution properties of griseofulvin-PEG ICs did not vary monotonically with the PEG MW, but rather an optimal intermediate PEG MW (i.e., 6 kDa) existed (Fig. 6). This is because of the griseofulvin ICs with too short or too long PEG chains possessed relatively higher crystal defects concentration (chain end defects and conventional crystal defects, respectively), and these ICs would exhibit faster transition to the pure griseofulvin crystals during dissolution, and consequently lost the IC dissolution advantage [34].

|
Download:
|
| Fig. 6. Concentration of dissolved griseofulvin with time in different griseofulvin solid forms. Adapted with permission [34]. Copyright 2016, Elsevier. | |
{kind=link}
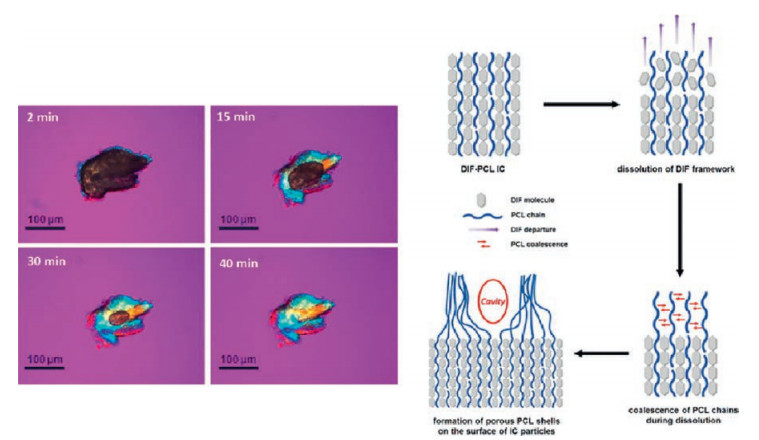
In contrast, when hydrophobic polymers were used as the guest, the drug-polymer ICs showed reduced dissolution rate and solubility than the original drug crystals [52]. More interestingly, compared with ICs with soluble PEG chains, drug-PCL ICs generally showed higher stability in the dissolution media and the residual crystals maintained the IC structure without dissociation. This demonstrates the possibility of using drug-polymer ICs to design and develop sustained-release drug products. In addition, since PCL is almost insoluble in water, the PCL chains initially residing in the host channels coalesced and formed crystalline but highly porous polymeric shells on the IC particles (Fig. 7). Ideally, these biodegradable PCL chains may form a continuous (instead of being porous) PCL shell on the drug particles. This core-shell structure would represent another novel drug solid forms, and its formation and potential applications deserve more studies in the future.

|
Download:
|
| Fig. 7. In-situ polarized optical microscope (POM) observation of the dissolution process of a diflunisal-PCL IC particle (left), and the proposed mechanism for the formation of porous PCL shell on the dissolving IC particles (right). Adapted with permission [52]. Copyright 2017, American Chemical Society.. | |
{kind=link}
In addition to the hydrophilic or hydrophobic homopolymers, amphiphilic block copolymers can also be used to tune the dissolution profiles of drug ICs. By changing the block lengths, the hydrophilicity/hydrophobicity of the guest polymers, as well as the host-guest interactions, can be adjusted in a controllable manner. For example, the dissolution behavior of diflunisal-PEG-b-PCL ICs showed a systematic variation with guest block lengths, where the equilibrium solubility and dissolution rate of the ICs crystals gradually decreased with increasing the PCL block length (the PEG block length remained a constant) [57]. In addition, in-situ formation of micelles during IC dissolution may also occur when using amphiphilic block copolymers as the guest, and its potential impact on the dissolution behaviors of drugs from IC crystals also deserves more studies.
4. SummaryFrom our studies described above and the previous work reported in the literature, the drug-polymer crystalline inclusion complexes have been established as a new pharmaceutical solid form. The crystal structures, as well as the general physicochemical properties of these ICs have been demonstrated with a few specific drug-polymer systems. In addition, a search strategy based on the formation of isostructural channel-type solvates was proposed to identify the drug candidates with the potential to form ICs with polymers.
In the future, the short list of the drug-polymer IC systems should be extended to more drug molecules beyond these model compounds. The formation of drug-polymer ICs provides more options to modulate the physicochemical properties of drug solids, and therefore help to develop drug products with optimized pharmaceutical profiles. ICs with hydrophilic polymer guests generally dissolve faster and have higher solubility than pure drug crystals. They can generate transient supersaturation in vivo and improve the drug oral bioavailability similar to drug co-crystals and drug/polymer solid dispersion. These drug-polymer crystalline ICs have similar structures to those drug solvate crystals, but have higher stability due to the much larger size of the polymer guests. On the other hand, the incorporation of hydrophobic polymers (e.g., PCL) in drug-polymer ICs has been proved effective in reducing the solubility and hence the dissolution rate of drugs in our studies, which exhibits great potential in the application of sustained-release drug systems. In addition, the in-situ formed core-shell structure during IC dissolution may represent another novel sub-category of drug solid forms with potential applications in long-acting injectables.
AcknowledgmentThis work was financially supported by the National Natural Science Foundation of China (No. 21434008).
| [1] |
C.C. Sun, J. Pharm. Sci. 98(2009) 1671-1687. DOI:10.1002/jps.21552 |
| [2] |
C.R. Gardner, C.T. Walsh, O. Almarsson, Nat. Rev. Drug Discov. 3(2004) 926-934. DOI:10.1038/nrd1550 |
| [3] |
H. Williams, N. Trevaskis, S. Charman, et al., Pharmacol. Rev. 65(2013) 315-499. DOI:10.1124/pr.112.005660 |
| [4] |
G. Bolla, A. Nangia, Chem. Commun. 52(2016) 8342-8360. DOI:10.1039/C6CC02943D |
| [5] |
S.R. Vippagunta, H.G. Brittain, D.J.W. Grant, Adv. Drug Deliv. Rev. 48(2001) 3-26. DOI:10.1016/S0169-409X(01)00097-7 |
| [6] |
A.D. Bond, Curr. Opin Solid State Mater. Sci. 13(2009) 91-97. DOI:10.1016/j.cossms.2009.06.004 |
| [7] |
J. Bernstein, Cryst. Growth Des. 11(2011) 632-650. DOI:10.1021/cg1013335 |
| [8] |
G.R. Desiraju, Cryst. Growth Des. 8(2008) 3-5. DOI:10.1021/cg701000q |
| [9] |
B.C. Hancock, G. Zografi, J. Pharm. Sci. 86(1997) 1-12. DOI:10.1021/js9601896 |
| [10] |
L. Yu, Adv. Drug Deliv. Rev. 48(2001) 27-42. DOI:10.1016/S0169-409X(01)00098-9 |
| [11] |
J.A. Baird, B. Van Eerdenbrugh, L.S. Taylor, J. Pharm. Sci. 99(2010) 3787-3806. DOI:10.1002/jps.22197 |
| [12] |
M.D. Ediger, P. Harrowell, J. Chem. Phys. 137(2012) 080901. DOI:10.1063/1.4747326 |
| [13] |
N. Schultheiss, A. Newman, Cryst. Growth Des. 9(2009) 2950-2967. DOI:10.1021/cg900129f |
| [14] |
C.C. Sun, Expert Opin. Drug Deliv. 10(2013) 201-213. DOI:10.1517/17425247.2013.747508 |
| [15] |
J.W. Steed, Trends Pharmacol. Sci. 34(2013) 185-193. DOI:10.1016/j.tips.2012.12.003 |
| [16] |
N.K. Duggirala, M.L. Perry, Ö. Almarsson, M.J. Zaworotko, Chem. Commun 52(2016) 640-655. DOI:10.1039/C5CC08216A |
| [17] |
E. Grothe, H. Meekes, E. Vlieg, J.H. Ter Horst, R. De Gelder, Cryst. Growth Des. 16(2016) 3237-3243. DOI:10.1021/acs.cgd.6b00200 |
| [18] |
A. Nangia, G.R. Desiraju, Chem. Commun. 7(1999) 605-606. |
| [19] |
R.K. Khankari, D.J.W. Grant, Thermochim. Acta 248(1995) 61-79. DOI:10.1016/0040-6031(94)01952-D |
| [20] |
A.T.M. Serajuddin, Adv. Drug Deliv. Rev. 59(2007) 603-616. DOI:10.1016/j.addr.2007.05.010 |
| [21] |
A. Avdeef, Adv. Drug Deliv. Rev. 59(2007) 568-590. DOI:10.1016/j.addr.2007.05.008 |
| [22] |
S.L. Childs, G.P. Stahly, A. Park, Mol. Pharm. 4(2007) 323-338. DOI:10.1021/mp0601345 |
| [23] |
G.P. Stahly, Cryst. Growth Des. 7(2007) 1007-1026. DOI:10.1021/cg060838j |
| [24] |
S.J. Dengale, H. Grohganz, T. Rades, K. Löbmann, Adv. Drug Deliv. Rev. 100(2016) 116-125. DOI:10.1016/j.addr.2015.12.009 |
| [25] |
K. Löbmann, C. Strachan, H. Grohganz, et al., Eur. J. Pharm. Biopharm. 81(2012) 159-169. DOI:10.1016/j.ejpb.2012.02.004 |
| [26] |
S.H. Lee, J.H. Bae, Y. Park, et al., Cryst. Growth Des. 15(2015) 3123-3130. DOI:10.1021/acs.cgd.5b00074 |
| [27] |
S. Yamamura, H. Gotoh, Y. Sakamoto, Y. Momose, Int. J. Pharm. 241(2002) 213-221. DOI:10.1016/S0378-5173(02)00195-3 |
| [28] |
S. Janssens, G. Van den Mooter, J. Pharm. Pharmacol. 61(2009) 1571-1586. DOI:10.1211/jpp.61.12.0001 |
| [29] |
Y. Huang, W. Dai, Acta Pharma. Sinica B 4(2014) 18-25. DOI:10.1016/j.apsb.2013.11.001 |
| [30] |
A.T.M. Serajuddin, J. Pharm. Sci. 88(1999) 1058-1066. DOI:10.1021/js980403l |
| [31] |
W.L. Chiou, S. Riegelmant, J. Pharm. Sci. 60(1971) 1281-1302. DOI:10.1002/jps.2600600902 |
| [32] |
Z. Zhong, C. Guo, L. Chen, J. Xu, Y. Huang, Chem. Commun. 50(2014) 6375-6378. DOI:10.1039/C4CC00159A |
| [33] |
Z. Zhong, C. Guo, X. Yang, et al., Cryst. Growth Des. 16(2016) 1181-1186. DOI:10.1021/acs.cgd.6b00010 |
| [34] |
X. Yang, Z. Zhong, Y. Huang, Int. J. Pharm. 508(2016) 51-60. DOI:10.1016/j.ijpharm.2016.05.014 |
| [35] |
K.D.M. Harris, Chem. Soc. Rev. 26(1997) 279-289. DOI:10.1039/cs9972600279 |
| [36] |
K.D.M. Harris, Supramol. Chem. 19(2007) 47-53. DOI:10.1080/10610270600977706 |
| [37] |
K.D.M. Harris, J.M. J. Chem. Soc. Thomas, Faraday Trans. 86(1990) 2985-2996.
|
| [38] |
A. Chenite, F. Brisse, Macromolecules 24(1991) 2221-2225. DOI:10.1021/ma00009a015 |
| [39] |
C. C. Sun, PCT Pat. Appl. (2006) 2006/024930 A1.
|
| [40] |
M. Dosiere, J. Macromol. Sci. Part B 35(1996) 303-328. DOI:10.1080/00222349608220383 |
| [41] |
G. Guerra, C. Daniel, P. Rizzo, O. Tarallo, J. Polym. Sci. Part B:Polym. Phys. 50(2012) 305-322. DOI:10.1002/polb.23035 |
| [42] |
G.R. Desiraju, Angew. Chem. Int. Ed. Eng. 34(1995) 2311-2327. DOI:10.1002/(ISSN)1521-3773 |
| [43] |
G.R. Desiraju, J. Am. Chem. Soc. 135(2013) 9952-9967. DOI:10.1021/ja403264c |
| [44] |
A. Mukherjee, Cryst. Growth Des. 15(2015) 3076-3085. DOI:10.1021/acs.cgd.5b00242 |
| [45] |
A.Y. Lee, D. Erdemir, A.S. Myerson, Annu. Rev. Chem. Biomol. Eng. 2(2011) 259-280. DOI:10.1146/annurev-chembioeng-061010-114224 |
| [46] |
B.G. Pereira, F.D. Fonte-Boa, J.A.L.C. Resende, et al., Cryst. Growth Des. 7(2007) 2016-2023. DOI:10.1021/cg0704495 |
| [47] |
M.J.L. Mariette, R.C. Mino, P.L. Antonie, G.D.V. Jakkie, J. Pharm. Sci. 76(1987) 744-752. DOI:10.1002/jps.2600760914 |
| [48] |
D. Swern, Ind. Eng. Chem. 47(1955) 216-221. DOI:10.1021/ie50542a023 |
| [49] |
G. Allegra, M. Farina, A. Immirzi, et al., J. Chem. Soc. B:Phys. Org.(1967), 1020-1028. DOI:10.1039/j29670001020 |
| [50] |
H.R. Allcock, Acc. Chem. Res. 11(1978) 81-87. DOI:10.1021/ar50123a001 |
| [51] |
H.R. Allcock, L.A. Siegel, J. Am. Chem. Soc. 80(1964) 5140-5144. |
| [52] |
Z. Zhong, X. Yang, B. Guo, J. Xu, Y. Huang, Cryst. Growth Des. 17(2017) 355-362. DOI:10.1021/acs.cgd.6b01578 |
| [53] |
D.J. Good, N. Rodri'guez-Hornedo, Cryst. Growth Des. 9(2009) 2252-2264. DOI:10.1021/cg801039j |
| [54] |
N.J. Babu, A. Nangia, Cryst. Growth Des. 11(2011) 2662-2679. DOI:10.1021/cg200492w |
| [55] |
R. Thakuria, A. Delori, W. Jones, et al., Int. J. Pharm. 453(2013) 101-125. DOI:10.1016/j.ijpharm.2012.10.043 |
| [56] |
S.L. Childs, L.J. Chyall, J.T. Dunlap, et al., J. Am. Chem. Soc. 126(2004) 13335-13342. DOI:10.1021/ja048114o |
| [57] |
Z. Zhong, X. Yang, X. Fu, et al., Chin. Chem. Lett. 28(2017) 1268-1275. DOI:10.1016/j.cclet.2017.04.001 |