 2017, Vol. 28
2017, Vol. 28 



b Key Laboratory of Polymer Chemistry & Physics of Ministry of Education, Center for Soft Matter Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
"Click" chemistry, along with its fundamental principles, has reshaped many research fields from materials science to biology ever since its conceptualization by Kolb et al. in 2001 [1]. The "click" philosophy embraces a set of nearly "ideal" chemical reactions, such as azide-alkyne Huisgen cycloaddition, that can join molecules together through carbon-heteroatom links in a highly modular and orthogonal fashion. It has been found to be particularly useful in polymer and materials science [2]. However, applying these"click" reactions to biological systems often requires extra chemical/enzymatic steps to introduce unnatural moieties to biomolecules, which could be troublesome for sensitive protein targets or living systems. Moreover, it remains a challenge to diversify "click" reactions in chemical space given the intrinsic difficulties in finding new reactions that meet the "click" criteria. On the other hand, the reactivities of biological motifs (e.g., natural amino acids) have been largely untapped in terms of expanding the territory of "click" chemistry. The gap remains to be filled at the cross-road of these two categories.
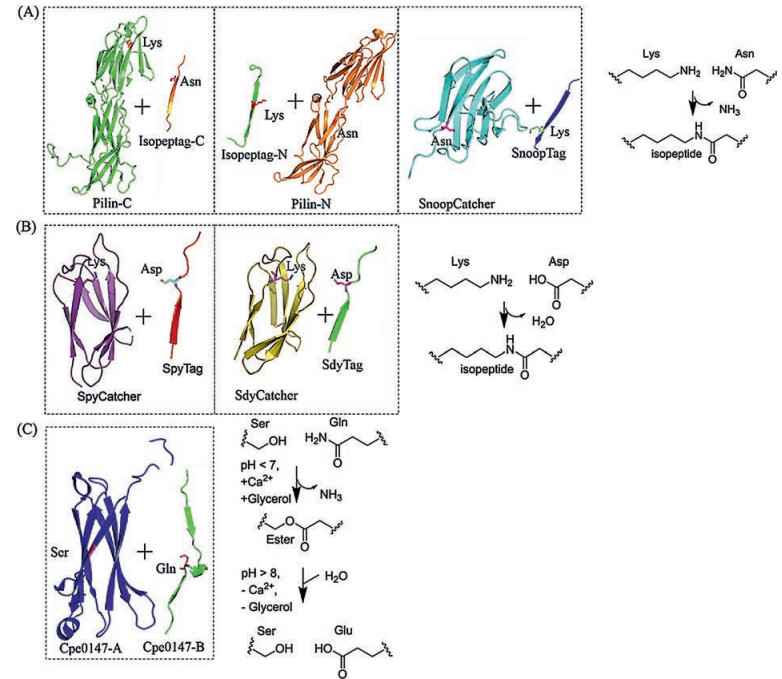
The recent discovery of naturally occurring, stabilizing isopeptide bonds in Gram-positive bacterial adhesins inspired the development of reactive peptide-protein partners [3]. To date, they include the first generation of pilin-C/isopeptag-C and pilinN/isopeptag-N [4], the gold standard SpyTag/SpyCatcher chemistry [5] and its homologous version, SdyTag/SdyCatcher reactive pair [6], and orthogonal version, SnoopTag/SnoopCatcher chemistry [7]. These reactive pairs are also known as "bacterial superglue" possessing infinite affinity to their cognate partners [8]. They have attracted significant attention and have been used in a variety of applications, ranging from materials science, macromolecular engineering, bio-imaging, protein purification to synthetic biology [9-14]. It is also possible to engineer ester-forming reactive pairs [15]. This type of protein chemistry is unique in that a single system integrates both molecular recognition and spontaneous isopeptide bond formation. Unlike split intein or sortase-based bioconjugation methods that can only take place in the termini of a protein or peptide [16, 17], they usually work anywhere on a protein (at N-, C-terminal, or internal sites). An additional extraordinary feature is that these peptide-protein reactions arefully genetically encodable [18]. Among them, SpyTag/SpyCatcher chemistry, developed by splitting the second immunoglobulin-like collagen adhesin domain (CnaB2) from the fibronectin binding protein (FbaB) of Streptococcus pyogenes [5], turns out to be the best of its kind, and itexhibits many features reminiscent of "click" reactions, including rapid kinetics, high yields, equimolarity, modularity, chemo-selectivity, and single reaction trajectory [18]. Hence, we conceive the concept of genetically encoded "click" chemistry (GECC) to describe a family of "ideal" peptide-protein reactive pairs that possesses the essential features of "click" chemistry while also being genetically encodable.
Similar to "click" chemistry in materials science, GECC is also a chemical philosophy referring to "perfect" peptide-proteinreactionswithall "ideal" features [2]. In reality, there may be nopeptide/ protein reactive pair that can meet all the requirements of GECC. Nevertheless, by illustrating the underlying principles and important criteriaof an ideal GECC, we hope to provoke the creativity from the community to develop new peptide-protein reactive pairs approximating the ideal GECC. We envision that GECC would add diverse features to the control of chemical reactivity through information transfer in materials and greatly expand the utility of this technology in biomaterials and synthetic biology, both in vivo and in vitro. Since there have already been a few excellent reviews on these "molecular superglues" and their broad applications [8-11], the focus of this mini-review would be on the prospectus of GECC in the context of materials science with the SpyTag/ SpyCatcher chemistry as a prime example.
2. SpyTag/SpyCatcher chemistry: the prototype of GECCAccording to Sharpless' definition [1], a click reaction must be modular, wide in scope, chemo-selective and must proceed by a single reaction trajectory. Additional requirements for a click reaction in the context of polymer science have also been proposed, which include equimolarity, large-scale purification, fast timescale, high yields and stable compounds [2]. The widely used Cu-catalyzed alkyne-azide cycloaddition (CuAAC) has thus far represented the most classic "click" reaction. A careful examination revealed that the SpyTag/SpyCatcherchemistry meets nearly all the click criteria mentioned above [2, 18], suggesting that this chemistry may usher in a new category of "click" chemistry with full genetic encodability and therefore can be regarded as a prototype of GECC. The SpyTag/SpyCatcher chemistry is highly selective, modular and broad in substratescope. Previous studies have shown that mixing equimolar SpyTag and SpyCatcher enabled a nearly quantitative conjugation of the two reactants with the only byproduct, H2O, under physiological conditions within a couple of hours [5, 18]. Zhang et al. [18] demonstrated that the SpyTag/SpyCatcher chemistry can work efficiently at N-and Cterminias well as in the middle of unstructured elastin-like polypeptides (ELPs). Schoene et al. [19] showed that SpyTag/ SpyCatcher can be genetically fused with either N-or C-terminus of a variety of globular proteins such as GFP, MBP, GST, and b-lactamase and retains their reactivities. Fast timescale is another prominent feature of the SpyTag/SpyCatcher chemistryof which thesecond-order rate constant wasdetermined to be ~1.4 ×103 L mol-1 s-1 [5]. This is substantially faster than the common CuAAC of which the second-order rate constants generally range from 10 L mol-1 s-1 to 200 L mol-1 s-1 [20]. Unlike many other biomacromolecular systems that are sensitive toward external conditions, the SpyTag/SpyCatcher reaction is robust and can tolerate a wide variety of buffers with different pH (5-8) and temperature (4-37 ℃) [5]. All of these are in agreement with the stringent demands of "click" chemistry. What distinguishes the SpyTag/SpyCatcher chemistry from traditional "click" chemistry is that protein folding is essential forits reaction. It does not tolerate protein denaturing conditions such as organic solvents (DMF, DMSO, etc.) and thus has limitations when used insome nonbiological systems. In addition, both reactants are also much bulkier than traditional click moieties like azide and alkyne, which may affect their reactivity in certain circumstances. Still, given the full genetic encodability and amicability toward biological systems, this prototype of GECC could serve as a surrogate superior to traditional click reactions in the context of proteinbased materials, particularly in cases where proteins are recalcitrant or sensitive to chemical modifications.
There are more that GECC canoffer. Diversification of the conventional "click" toolbox is nontrivial given the inherent difficulties in finding new "good" reactions in chemical space that meet the "click" criteria. By contrast, the genetic encodability of GECC makes it possible to develop other reaction pairs with alteredspecificity even based on the same reaction type. In principle, orthogonal GECC pairscould be developed by splitting and engineering naturally occurringisopeptide-containing protein domains [7] or derived fromexisting reactive pairs through directed evolution [22]. Since the spontaneously formedisopeptide bond is a common structural motif existing in Grampositive bacterial pilus proteins [23], the GECC pairs could emerge from these natural protein domains. Once a basal-level reactivity is identified, appropriate protein engineering may be applied to improve the reactivity of these protein/peptide pairs to meet the click criteria. Several primitive protein/peptide molecular superglues that reconstitute and spontaneously form covalent bonds have been developed in the past years from the earliest examples of pilin-C/isopeptag-C and pilin-N/isopeptagN [4] to the later SpyTag/SpyCatcher [5] and SnoopTag/ SnoopCatcher reactive pairs [7] (Fig. 1). The first = generationsuperglues, pilin-C/isopeptag-C and pilin-N/isopeptag-N, suffered from several shortcomings including slow reaction kinetics, low reconstitution efficiency and large size of the reactants. Thus, strictly speaking, these earlyversionsof molecular superglues are not GECC andless appealing for practical applications. Although the split fragments initially derived from Streptococcus pyogenes fibronectin-binding protein, FbaB, also exhibited a poor reconstitution efficiency, Zakeri et al. [5] were able to improve the reactivity through intense protein engineering, which ultimately led to the creation of highly efficient SpyTag/ SpyCatcher chemistry with "click" features. The superglue chemistry gradually gains popularity ever since.

|
Download:
|
| Fig. 1. Superglue-like protein/peptide pairs that spontaneously form covalent bonds. (A) Lys-Asnisopeptide-based superglue pairs, pilin-C/isopeptag-C, pilin-N/isopeptag-N (PDB code 3B2M) and SnoopCatcher/SnoopTag (PDB code 2WW8). (B) Lys-Asp isopeptide-based superglue pairs, SpyCatcher/SpyTag (PDB code 4MLI) and SdyCatcher/SdyTag (Swiss-Model modeling). (C) Reversible Ser-Gln ester-bond-based superglue pair, Cpe0147-A/Cep0147-B (PDB code 4MKM). | |
3. The state-of-the-art toolbox toward GECC
Following the same strategy, an alternative isopeptide-bond forming SnoopTag/SnoopCatcherpair was developed by splitting another adhesin, RrgA, from Streptococcus pneumoniae (Fig. 1A) [7]. The resulting SnoopTag/SnoopCatcher chemistry is orthogonal to the existing SpyTag/SpyCatcher chemistry and shows no crossreactivity. But itsefficiency is not as great as theSpyTag/SpyCatcher chemistry. Tan et al. [6] developed a similar isopeptide bondforming pair, SdyTag/SdyCatcher, by splitting a Streptococcus dysgalactiae fibronectin binding protein (Fig. 1B). Owing to their homology, the SdyTag/SdyCatcher chemistry exhibited residual cross-reactivity toward the SpyTag/SpyCatcher. In addition to the isopeptide-based protein/peptide pairs, a recent study demonstrated the development of a Thr-Gln ester-bond-forming protein/ peptide pair by dissecting a Clostridium perfringens adhesin Cpe0147 (Fig. 1C) [15]. Unlike the irreversible isopeptide-based molecular superglues, the reaction of Cpe0147-A/Cpe0147-B is sensitive to pH; the ester bond forms under low pH conditions (pH < 7) and can subsequently be hydrolyzed under basic conditions (pH > 8). This pH-switchable reactive pair offers the possibility of creating flexible protein architectures that can be assembled and disassembled in a pH-dependent manner.
A unique feature of the GECC is that a single system integrates structure (sequence)-based molecular recognition, which dictates specificity, and the subsequent covalent bond formation, which determines the reaction mode. Such a dual interaction mode offers a great leverage to engineer and control these reactions. In principle, orthogonal GECCs can be readily developed by targeting the molecular recognition while preserving the chemistry within the reaction pocket through directed evolution. Numerous additional features could be programmed into the sequence to make it responsive to environmental stimuli such as pH, temperature, light and metal ions so that the chemistry can be controlled via these stimuli. As proof of concept, it has been shown that through extensive mutations, SpyCatcher can be negatively supercharged to become an intrinsically disordered polypeptide, SpyCatcher (-), that exhibits decent and stimuli-responsive chemical reactivity toward wild-type SpyTag [32]. Even for the same structural scaffold like SpyTag-SpyCatcher reactive pair, distinct reactivity could be programed with as few as three mutations [22]. Interestingly, the catalytic triad consisting of LysAsp (or Asn)-Glu conserved in molecular supergluesturns outto be robust and can be "written" artificially into other sequences as well. Kwon et al. [33] reported encouragingly the de novo design of an isopeptide bond into an immunoglobulin-like protein domain, which raises the possibility of rationally introducing artificial isopeptide bonds into bio-macromolecules or even unnatural molecular scaffolds. Technologies such as phage display or yeast/ bacterial cell surface display can also be used to screen and identify new reactive pairs. It is also desirable to have GECCs comprising smaller protein/peptide reactants, which may enable less-trace bio-molecular labeling in vitro and in vivo. Admittedly, these pairs to date are generally less reactive than SpyTag/SpyCatcher and could hardly be qualifiedas GECC. Further protein engineering is needed to improve their reactivity to a level comparable to that of typical click reactions. It is even possible that their reactivity (such as that of SpyTag/SpyCatcher chemistry) can be elevated to beat the diffusion limit (~109 L mol-1 s-1). We anticipate that a palette of such tools with diverse features will be created for the research community, reminiscent of the epic of fluorescent proteins.
4. Protein topology engineeringAn immediate application of GECC is to program the posttranslational modification of proteins for topology engineering. Most biomacromolecules, such as DNA, RNA or proteins, are linear biopolymers by nature. Proteins and RNAs are capable of forming three-dimensional structures through folding, which is mainly driven by noncovalent interactions such as hydrophobic interactions, hydrogen bonding, van der Waal interactions, electrostatic interactions, etc. Expanding biopolymer topology beyond linear configuration using cellular machinery may lead to new materials with properties that have never been explored before. GECC and other superglue technologieshave been demonstrated to be extremely useful in this regard [18]. Shortly after the development of SpyTag/SpyCatcher chemistry, Zhang et al. [18] demonstrated cellular synthesis of cyclic and tadpole-shaped elastin-like proteins (ELPs) as well as in vitro assembly of ELPs into other non-linear topologies such as block, 3-arm star, 4-arm star and Hshaped proteins. Using globularproteins such as β-lactamase, dihydrofolate reductase, phytase, firefly luciferase and lichenase as model systems, subsequent studies showed that protein cyclization ("SpyRing") enabled by the SpyTag/SpyCatcher chemistry significantly improved the protein stability toward heat and proteolysis [24-27]. In combination with dimeric p53 domain, the SpyTag/SpyCatcher chemistry also enabled Zhang and co-workers [28, 29] to create protein catenanes and related topologies. They further showed that the formation of protein catenanes provided a convenient and versatile approach to stabilize the protein of interest via topology engineering [28]. A recent study investigated on the influence of different topological structures on the phase transition of ELPs, pointing to a new way to tune the physical properties of ELPs [30]. Table 1 summarizes the examples ofprotein topology engineering using SpyTag/SpyCatcher chemistry.
|
|
Table 1 Topology engineering enabled by SpyTag/SpyCatcher chemistry. |

{kind=link}
5. Entirely genetically engineered protein-based hydrogels
A central question facing thebottom-up approach for material design is how to transfer the function at the molecular level to the material properties at a macroscopic level [35-37]. The last decade has witnessed a growing trend of designing materials with dynamically tunable properties [38]. These "smart" materials necessitate a new level of control over the structural and functional properties of macromolecules as well as their interactions with external stimuli. Natural evolution has led to the creation of a vast number of protein molecules with extraordinary structural and functional diversity, which have long served as a source of inspiration for designing and synthesizing new molecular entities and materials [39-41]. Many of these bio-macromolecules are also known to be able to sense and respond to environmental stimuli such as light, pH, temperature, metal ions, organic smallmolecules, which play essential roles in biological signaling and regulation [42]. Such an ecological diversity has yet to be fully utilized to design and create functional materials with dynamic properties.
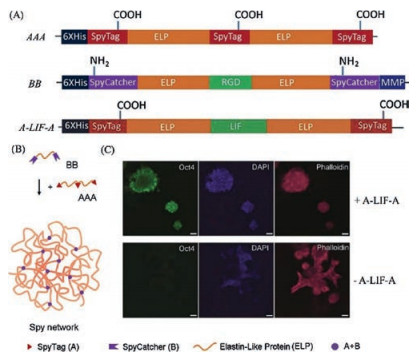
Many chemical methods conventionally used for polymerization or molecular assembly are incompatible with protein molecules that are labile upon chemical modification. In light of its high efficiency and specificity, the SpyTag/SpyCatcher chemistry offers a versatile approach for covalently stitching together recombinant proteins to form a polymeric protein network under mild physiological conditions. Sun et al. [21] demonstrated the success of synthesizing an entirely recombinant ELP-based hydrogel, namely the "Spy network", using the SpyTag/SpyCatcher chemistry, which can further be designed to contain multiple bioactive motifs such as RGD cell binding domains, matrix metalloproteinase (MMP) cleavage sites, and even leukemia inhibitory factors (LIFs) (Fig. 2). The resulting LIF-Spy network hydrogel has been shown to be able to support 3D culturing of mouse embryonic stem cells (mESCs) (Fig. 2C). Gao et al. [31] also used the SpyTag/SpyCatcher chemistry to synthesize globular protein (GB1) -based hydrogels with excellent biocompatibility. These Spy network hydrogels demonstrated the feasibility of directly assembling recombinant protein molecules intomacroscopic materials with no need of any chemical modification while maintaining the molecular functions within the materials' scaffolds.

|
Download:
|
| Fig. 2. Synthesis of bioactive protein hydrogels using SpyTag/SpyCatcher chemistry. (A) Genetic constructs for protein building blocks used for the synthesis of LIF-Spy network hydrogels. (B) Schematic illustration of the Spy network hydrogel formed by the reaction of AAA + BB. (C) The Spy network hydrogel containing A-LIF-A supports the pluripotency of mouse embryonic stem cells (mESCs). Reprinted with permission [21]. | |
{kind=link}
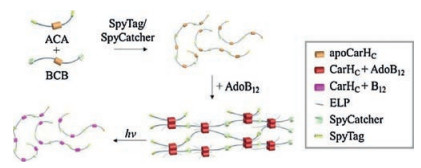
Dynamic, stimuli-responsive biomaterials hold great promise for stem cell regulation as well as therapeutic delivery [45]. Given the great diversity of stimuli-responsive proteins in nature, the direct assembly of these proteins into macroscopic materials would provide a versatile approach for developing "smart" materials. The challenge lies in how to faithfully transfer the molecular functions to the resulting material properties through the polymerization/assembly process. Taking advantage of the SpyTag/SpyCatcher chemistry, Wang et al. [34] recently created B12-dependent photo-responsive, entirely protein-based hydrogels by covalently polymerizing light-sensitive CarH proteins. The CarH protein has been known to be an adenosinecobalamine (AdoB12)-binding light-dependent transcriptional regulator that controls carotenoid biosynthesis in bacterial cells [46-49]. A unique feature of CarH is that its C-terminal domains (CarHC) form a tetramer when binding to thecofactor AdoB12 under dark conditions and disassemble into monomers accompanied with the photolysis of AdoB12upon exposure to green light (512 nm) or white light. CarHCpolymers that were synthesized simply by mixing telechelicSpyTag-ELP-CarHC-ELP-SpyTag (ACA) and SpyCatcher-ELPCarHC-ELP-SpyCatcher (BCB) at an equimolar ratio formed a hydrogel in the presence of AdoB12 in the dark and underwent a rapid gel-sol transition upon exposure to light (Fig. 3). Such a lightdependent gel-sol transition was utilized for the controlled release of stem cells and protein molecules in vitro [34]. Together, the results point to a powerful methodology based on GECC for directly converting stimuli-responsive proteins into functional materials with dynamic properties.

|
Download:
|
| Fig. 3. Synthesis of a photo-responsive CarHC hydrogel by SpyTag/SpyCatcher chemistry. Reprinted with permission [34]. | |
{kind=link}
6. Assembly of protein nanomaterials for biomedical applications
In addition to macroscopic soft materials, the newly developed bacterial superglueshave also enabled the precise synthesis of advanced nanoarchitectures. Fairhead et al. [43] used a combination of the SpyTag/SpyCatcher chemistry and the ultrastable noncovalent biotin-traptavidin (a streptavidin variant with enhanced biotin binding affinity) interaction to create robust nanoassemblies including chimeric SpyAvidin tetramers (4 subunits), octamers (8 subunits) and eicosamers (20 subunits). Moreover, they further demonstrated that the SpyAvidineicosamers (Fig. 4) that displayed major histocompatibility complex (MHC) clusters could serveas a more powerful stimulus for T cell signaling than conventional MHC tetramers.

|
Download:
|
| Fig. 4. Generation of a SpyAvidineicosamer (20 subunits) clustering trimeric major histocompatibility complex (MHC) class Ⅰ:β2-microglobulin:peptide complexes through a combination of noncovalent biotin/trapatavidin and covalent SpyTag/ SpyCatcher interactions. The resulting MHC eicosamers served as a powerful stimulus to T cell signaling. Reprinted with permission [43]. Copyright 2014, American Chemical Society. | |
{kind=link}
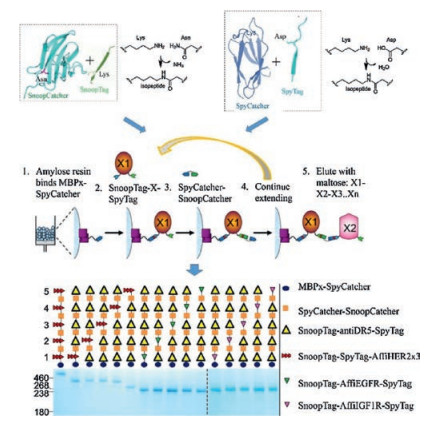
Combining the two orthogonal superglue pairs (SpyTag/ SpyCatcher and SnoopTag/SnoopCatcher) in solid-phase synthesis, Veggiani et al. [7] recently achieved the sequence-programmed synthesis of polyprotein teams ("polyproteams") containing different kinds of protein building blocks, which provides an access to a new area of biological space (Fig. 5). They further showed the synthesis of a combinatorial library of polyproteams containing repeats of bioactive protein molecules including a nanobody agonist for Death Receptor 5 (DR5) and an affibody to either epidermal growth factor receptor (EGFR), HER2, or type Ⅰ insulin-like growth factor receptor (IGF1R). Screening the polyproteam library eventually led to the finding of an optimal combination of these protein molecules for cell death induction in cancer cells.

|
Download:
|
| Fig. 5. Solid-phase polyproteam synthesis by two orthogonal superglue pairs, SpyTag/SpyCatcher and SnoopTag/SnoopCatcher. Reprinted with permission [7]. | |
{kind=link}
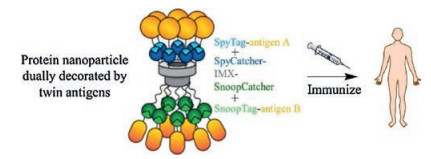
Synthetic vaccines represent the trend of future vaccine development because of their better safety and specificity compared with traditional vaccines based on weakened or killed microorganisms. The molecular superglues provide a rapid and versatile approach to generate synthetic vaccines through protein assembly. Liu et al. [50] demonstrated the success of synthesizing a vaccine by covalently stitching together dendritic cell-targeting molecules and specific model antigens using the SpyTag/ SpyCatcher chemistry. The resulting entirely protein-based vaccine preserved the individual function of different subunit proteins and exhibited the ability to induce T and B cell responses. Brune et al. [51] genetically engineered virus-like particles (VLPs) to display SpyCatcher on their surfaces, which further enabled the decoration of VLPs with SpyTag-containing malarial antigens. These VLPantigen conjugates were able to induce antibody response efficiently after only a single immunization, showcasing the potential of this simple, efficient and modular decoration method for vaccine development [52-55]. Brune et al. [44] also created an entirely protein-based assembly using the two orthogonal superglue pairs, SpyTag/SpyCatcher and SnoopTag/SnoopCatcher, to display dual malarial antigens, Pfs25 and Pfs28, on a single nanoparticle, which led to enhanced antibody response compared with monomeric proteins (Fig. 6). This multiple-antigen-display platform enabled by orthogonal superglues may provide a powerful weapon for combating malaria and other infectious diseases.

|
Download:
|
| Fig. 6. A multiple-antigen-display synthetic vaccine enabled by orthogonal superglues. Adapted and reprinted with permission [44]. Copyright 2017, American Chemical Society. | |
{kind=link}
7. Assembly of proteinnano-reactors
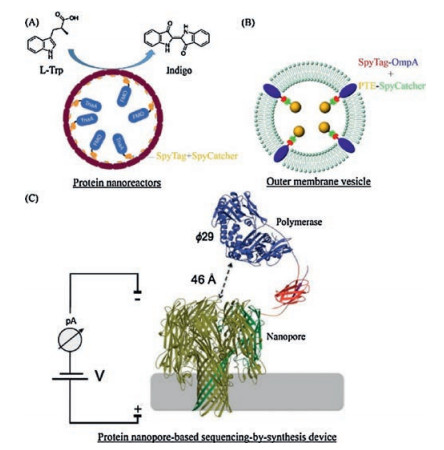
Compartmentalization of enzymes has been essential to some natural metabolic processes such as CO2 fixation by bacterial carboxysomes. These protein-based bacterial organelles promote a cascade of biochemical reactions by encapsulating and colocalizing enzymes with low-molecular-weight or even gaseous substrates and cofactors, by protecting cells from toxic intermediates, and by stabilizing vulnerable enzymes/intermediates in a confined microenvironment [56]. Inspired by these natural protein microcompartments, Giessen and Silver [57] created artificial MS2 phase capsid protein-basednano-compartments in Escherichia coli, where the interior surfaces were genetically engineered to be tagged with SpyTag for covalently encapsulatingSpyCatcher-fusion cargo proteins such as the enzymes involved in the indigo biosynthetic pathway (Fig. 7A). The resulting enzymatic nano-reactors not only led to an increased indigo production in vivo but also exhibited enhanced stability compared with free enzymes in vitro. In addition to the assembly of entirely protein-based nanoreactors, Alves et al. [58] demonstrated the success of directly packaging phosphotriesterase into bacterial outer membrane vesicles (OMVs) by taking advantage of SpyTag/ SpyCatcher chemistry and the outer membrane protein A (OmpA)-based bacterial outer membrane display system (Fig. 7B). The resultant enzymatic nano-proteoliposomes exhibited native-like enzyme kinetics and enhanced enzyme stability. Using SpyTag/ SpyCatcher chemistry, Stranges et al. [59] developed a nanoporebased sequencing-by-synthesis (Nanopore-SBS) technology by covalently stitching a DNA polymerase with a nanopore construct composed of an α-hemolysinheptamer (Fig. 7C). The technology, in combination with electrode arrays, paves the way for an economical, high-throughput, single-molecule, electronic sequencing platform. Recently, the high affinity of SpyTag/SpyCatcher chemistry has also been used to promote the efficiency of sortase-mediated ligation, suggesting its broad utility in proximity-enhanced interactions [60].

|
Download:
|
| Fig. 7. Assembly of protein nano-reactors by SpyTag/SpyCatcher chemistry. (A) An MS2 phase capsid protein-based nano-compartment assembled in E. coli. The interior surface was genetically engineered to be SpyTagged to covalently encapsulate the enzymes involved in the indigo biosynthetic pathway. The image was adapted with permission from Ref. [55]. Copyright 2016, John Wiley and Sons. (B) Cellular synthesis of enzymatic nano-proteoliposomes comprising phosphotriesterases (PTEs) and bacterial outer membrane vesicles enabled bya combination of Spy/Tag/SpyCatcher chemistry and bacterial OmpA system. The image was adapted with permission from Ref. [58]. Copyright 2015, American Chemical Society. (C) A nanopore-based sequencing-by-synthesis device created by covalently coupling a DNA polymerase to an α-hemolysinheptamer using the SpyTag/SpyCatcher chemistry.Reprinted with permission [59]. | |
{kind=link}
8. Conclusions
In summary, we put forth the idea of GECC to describe a category of "perfect" protein/peptide reactions and used SpyTag/ SpyCatcher chemistry as a prototype of GECC to illustrate their robustness for bio-macromolecular assembly, plasticity for engineering, and versatility for applications. The GECC represents a philosophical thinking in the context of protein science that depictsthe characteristics of an ideal genetically encoded protein reaction. Although few reactive pairs would suffice these stringent demands, the discussion of GECC per se opens the door to the development of new chemical tools that entailfully genetically encoded reactants and introduces a new dimension toprotein engineering and synthetic biology. Thanks to the great efforts made by chemists and protein engineers, the GECC toolbox will continue to expand beyond the current gold standard, SpyTag/ SpyCatcher chemistry. We believe that it will play increasingly important rolesin both fundamental biological studies and applied biotechnology in the near future. Although the concept of GECC is still in its infancy, there is new knowledge to be gained and new opportunities to be seized. The dawn of a new era awaits upon the unleashing of the chemical power from the vast protein sequence space.
AcknowledgmentsWe thank the financial supports from the Research Grants Council of Hong Kong SAR Government to F. Sun (RGC-ECS Nos. #26103915 and AoE/M-09/12) and from the 863 Program (No. 2015AA020941), the National Natural Science Foundation of China (Nos. 21474003, 91427304) and "1000 Plan (Youth)" to W. B. Zhang. In addition, F. Sun is grateful to the Department of Chemical and Biological Engineering, HKUST for the faculty start-up fund.
| [1] |
H.C. Kolb, M.G. Finn, K.B. Sharpless, Angew. Chem. Int. Ed. 40(2001) 2004-2021. DOI:10.1002/(ISSN)1521-3773 |
| [2] |
C. Barner-Kowollik, F.E. Du Prez, P. Espeel, et al., Angew. Chem. Int. Ed. 50(2011) 60-62. DOI:10.1002/anie.v50.1 |
| [3] |
H.J. Kang, F. Coulibaly, F. Clow, T. Proft, E.N. Baker, Science 318(2007) 1625-1628. DOI:10.1126/science.1145806 |
| [4] |
B. Zakeri, M. Howarth, J. Am. Chem. Soc. 132(2010) 4526-4527. DOI:10.1021/ja910795a |
| [5] |
B. Zakeri, J.O. Fierer, E. Celik, et al., Proc.Natl. Acad. Sci. U. S. A. 109(2012) E690-E697. DOI:10.1073/pnas.1115485109 |
| [6] |
L.L. Tan, S.S. Hoon, F.T. Wong, PLoS One 11(2016) e0165074. DOI:10.1371/journal.pone.0165074 |
| [7] |
G. Veggiani, T. Nakamura, M.D. Brenner, et al., Proc. Natl. Acad. Sci. U. S. A. 113(2016) 1202-1207. DOI:10.1073/pnas.1519214113 |
| [8] |
G. Veggiani, B. Zakeri, M. Howarth, Trends Biotechnol. 32(2014) 506-512. DOI:10.1016/j.tibtech.2014.08.001 |
| [9] |
S.C. Reddington, M. Howarth, Curr. Opin. Chem. Biol. 29(2015) 94-99. DOI:10.1016/j.cbpa.2015.10.002 |
| [10] |
M. Proschel, R. Detsch, A.R. Boccaccini, U. Sonnewald, Front Bioeng. Biotechnol. 3(2015) 168. |
| [11] |
B. Zakeri, ChemBioChem 16(2015) 2277-2282. DOI:10.1002/cbic.v16.16 |
| [12] |
C.N. Bedbrook, M. Kato, S. Ravindra Kumar, et al., Chem. Biol. 22(2015) 1108-1121. DOI:10.1016/j.chembiol.2015.06.020 |
| [13] |
H. Moon, Y. Bae, H. Kim, S. Kang, Chem. Commun. 52(2016) 14051-14054. DOI:10.1039/C6CC07363H |
| [14] |
V. Pessino, Y.R. Citron, S. Feng, B. Huang, ChemBioChem 18(2017) 1492-1495. DOI:10.1002/cbic.v18.15 |
| [15] |
P.G. Young, Y. Yosaatmadja, P.W.R. Harris, et al., Chem. Commun. 53(2017) 1502-1505. DOI:10.1039/C6CC09899A |
| [16] |
N.H. Shah, T.W. Muir, Chem. Sci. 5(2014) 446-461. DOI:10.1039/C3SC52951G |
| [17] |
M. Schmidt, A. Toplak, P.J. Quaedflieg, T. Nuijens, Curr. Opin. Chem. Biol. 38(2017) 1-7. |
| [18] |
W.B. Zhang, F. Sun, D.A. Tirrell, F.H. Arnold, J. Am. Chem. Soc. 135(2013) 13988-13997. DOI:10.1021/ja4076452 |
| [19] |
C. Schoene, J.O. Fierer, S.P. Bennett, M. Howarth, Angew. Chem. Int. Ed. 53(2014) 6101-6104. DOI:10.1002/anie.201402519 |
| [20] |
C.S. McKay, M.G. Finn, Chem. Biol. 21(2014) 1075-1101. DOI:10.1016/j.chembiol.2014.09.002 |
| [21] |
F. Sun, W.B. Zhang, A. Mahdavi, F.H. Arnold, D.A. Tirrell, Proc. Natl. Acad. Sci. U. S. A. 111(2014) 11269-11274. DOI:10.1073/pnas.1401291111 |
| [22] |
Y. Liu, D. Liu, W. Yang, et al., Chem. Sci. 8(2017) 6577-6582. DOI:10.1039/C7SC02686B |
| [23] |
U. Schwarz-Linek, M.J. Banfield, Proc. Natl. Acad. Sci. U. S. A. 111(2014) 1229-1230. DOI:10.1073/pnas.1322482111 |
| [24] |
C. Schoene, J.O. Fierer, S.P. Bennett, M. Howarth, Angew. Chem. Int. Ed. 53(2014) 6101-6104. DOI:10.1002/anie.201402519 |
| [25] |
C. Schoene, S.P. Bennett, M. Howarth, Sci. Rep. 6(2016) 21151. DOI:10.1038/srep21151 |
| [26] |
M. Si, Q. Xu, L. Jiang, H. Huang, PLoS One 11(2016) e0162318. DOI:10.1371/journal.pone.0162318 |
| [27] |
J.D. Wang, Y.L. Wang, X.Z. Wang, et al., Biotechnol. Biofuels 9(2016) 79. DOI:10.1186/s13068-016-0490-5 |
| [28] |
D. Liu, W.H. Wu, Y.J. Liu, et al., ACS Cent. Sci. 3(2017) 473-481. DOI:10.1021/acscentsci.7b00104 |
| [29] |
X.W. Wang, W.B. Zhang, Angew. Chem. Int. Ed. 55(2016) 3442-3446. DOI:10.1002/anie.201511640 |
| [30] |
D.D. Zhang, Z. Cai, J. Wang, et al., Sci. Sin. Chim. 46(2016) 881-890. |
| [31] |
X.Y. Gao, J. Fang, B. Xue, L.L. Fu, H.B. Li, Biomacromolecules 17(2016) 2812-2819. DOI:10.1021/acs.biomac.6b00566 |
| [32] |
Y. Cao, D. Liu, W. Zhang, Chem. Commun. 53(2017) 8830-8833. DOI:10.1039/C7CC04507G |
| [33] |
H. Kwon, P.G. Young, C.J. Squire, E.N. Baker, Sci. Rep. 7(2017) 42753. DOI:10.1038/srep42753 |
| [34] |
R. Wang, Z. Yang, J. Luo, I.M. Hsing, F. Sun, Proc.Natl. Acad.Sci. U. S. A. 114(2017) 5912-5917. DOI:10.1073/pnas.1621350114 |
| [35] |
W.B. Zhang, S.Z.D. Cheng, Chin. J. Polym. Sci. 33(2015) 797-814. DOI:10.1007/s10118-015-1653-8 |
| [36] |
W.B. Zhang, X. Yu, C.L. Wang, et al., Macromolecules 47(2014) 1221-1239. DOI:10.1021/ma401724p |
| [37] |
W.B. Zhang, X.M. Wang, X.W. Wang, et al., Prog. Chem. 27(2015) 1333-1342. |
| [38] |
J.A. Burdick, W.L. Murphy, Nat. Commun. 3(2012) 1269. DOI:10.1038/ncomms2271 |
| [39] |
W.B. Zhang, X.L. Wu, G.Z. Yin, Y. Shao, S.Z.D. Cheng, Mater. Horiz. 4(2017) 117-132. DOI:10.1039/C6MH00448B |
| [40] |
G.Z. Yin, W.B. Zhang, S.Z.D. Cheng, Sci. China Chem. 60(2017) 338-352. DOI:10.1007/s11426-016-0436-x |
| [41] |
W.B. Zhang, E.Q. Chen, J. Wang, et al., Acta Phys. Sin. 65(2016) 183601. |
| [42] |
R.H. Crabtree, Science 266(1994) 1591-1592. DOI:10.1126/science.266.5190.1591 |
| [43] |
M. Fairhead, G. Veggiani, M. Lever, et al., J. Am. Chem. Soc. 136(2014) 12355-12363. DOI:10.1021/ja505584f |
| [44] |
K.D. Brune, C.M. Buldun, Y.Y. Li, et al., Bioconjug. Chem. 28(2017) 1544-1551. DOI:10.1021/acs.bioconjchem.7b00174 |
| [45] |
R. Langer, D.A. Tirrell, Nature 428(2004) 487-492. DOI:10.1038/nature02388 |
| [46] |
R.J. Kutta, S.J. Hardman, L.O. Johannissen, et al., Nat. Commun. 6(2015) 7907. DOI:10.1038/ncomms8907 |
| [47] |
M. Jost, J.J. Fernandez-Zapata, M.C. Polanco, et al., Nature 526(2015) 536-541. DOI:10.1038/nature14950 |
| [48] |
J.M. Ortiz-Guerrero, M.C. Polanco, F.J. Murillo, S. Padmanabhan, M. EliasArnanz, Proc. Natl. Acad. Sci. U. S. A. 108(2011) 7565-7570. DOI:10.1073/pnas.1018972108 |
| [49] |
M. Jost, J.H. Simpson, C.L. Drennan, Biochemistry 54(2015) 3231-3234. DOI:10.1021/acs.biochem.5b00416 |
| [50] |
Z.D. Liu, H. Zhou, W.J. Wang, et al., Sci. Rep. 4(2014) 7266. |
| [51] |
K.D. Brune, D.B. Leneghan, I.J. Brian, et al., Sci. Rep. 6(2016) 19234. DOI:10.1038/srep19234 |
| [52] |
S. Thrane, C.M. Janitzek, S. Matondo, et al., J. Nanobiotechnol. 14(2016) 30. DOI:10.1186/s12951-016-0181-1 |
| [53] |
C.M. Janitzek, S. Matondo, S. Thrane, et al., Malar. J. 15(2016) 545. DOI:10.1186/s12936-016-1574-1 |
| [54] |
S.K. Singh, S. Thrane, C.M. Janitzek, et al., Vaccine 35(2017) 3726-3732. DOI:10.1016/j.vaccine.2017.05.054 |
| [55] |
D.B. Leneghan, K. Miura, I.J. Taylor, et al., Sci. Rep. 7(2017) 3811. DOI:10.1038/s41598-017-03798-3 |
| [56] |
C.A. Kerfeld, S. Heinhorst, G.C. Cannon, Annu. Rev. Microbiol. 64(2010) 391-408. DOI:10.1146/annurev.micro.112408.134211 |
| [57] |
T.W. Giessen, P.A. Silver, ChemBioChem 17(2016) 1931-1935. DOI:10.1002/cbic.v17.20 |
| [58] |
N.J. Alves, K.B. Turner, M.A. Daniele, et al., ACS Appl. Mater. Interface 7(2015) 24963-24972. DOI:10.1021/acsami.5b08811 |
| [59] |
P.B. Stranges, M. Palla, S. Kalachikov, et al., Proc. Natl. Acad. Sci. U. S. A. 113(2016) E6749-E6756. DOI:10.1073/pnas.1608271113 |
| [60] |
H.H. Wang, B. Altun, K. Nwe, A. Tsourkas, Angew. Chem. Int. Ed. 56(2017) 5349-5352. DOI:10.1002/anie.201701419 |