 2017, Vol. 28
2017, Vol. 28 



b School of Chemical Engineering, Sichuan University, Chengdu 610065, China;
c College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325027, China
Synergistic cancer therapy had attracted more and more attention for high efficient cancer therapy [1-4]. The theranostic nanoparticles (NPs) integrated with diagnostic imaging and therapeutic capabilities to realize imaging-guided drug delivery and tumor treatment [5, 6]. The platform combined with multiply treatments avoided drug resistance comparing to single chemotherapy [7, 8]. In recent years, the development of photodynamic therapy (PDT) generating reactive oxygen species (ROS) including singlet oxygen (1O2) and oxygen radicals became an effective cancer treatment [9-12]. The combination of photodynamic therapy and chemotherapy had exhibited promising effect on cancer therapy [13, 14]. Most photosensitizers in PDT were hydrophobic molecules and inconvenient for application. NPs encapsulation was an important way to improve the water solubility and tumor tissue specificity of photosensitizers [15, 16]. Another effect strategy to enhance the solubility of photosensitizers was preparing photosensitizers-polymer conjugate [1, 17, 18].
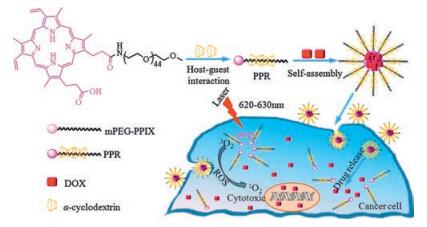
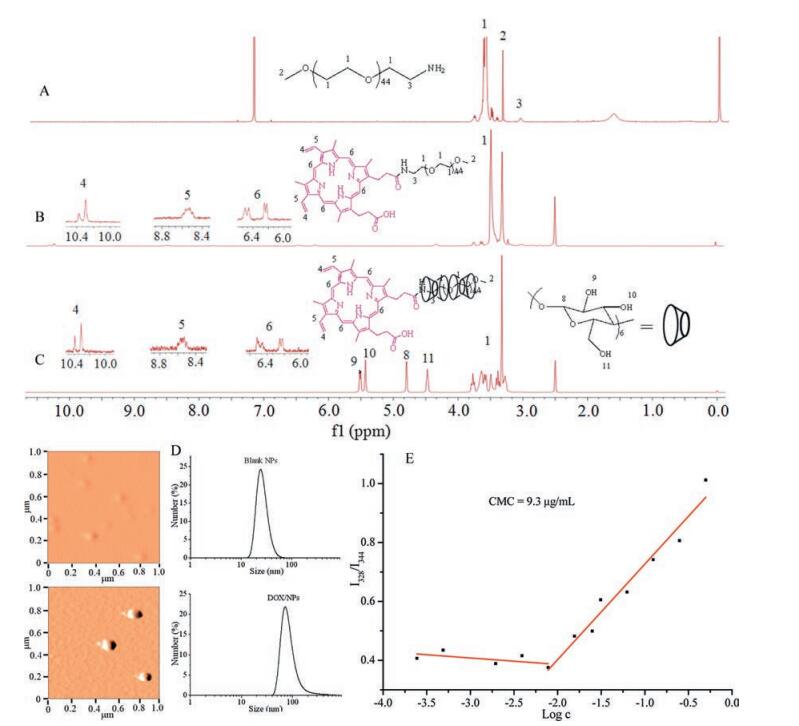
Polypseudorotaxane (PPR) composed of cyclic molecules and axis and end-capped by bulky groups at the terminal, had been widely used in molecule machine, drug controlled release, tissue engineering and so on, for its low toxicity, degradability, controllable size and unique architecture [19-24]. The α-cyclodextrins (α-CDs) had a hydrophobic cavity [25], and the diameter of the cavity matched the poly (ethylene glycol) (PEG) chain well, the α-CDs and PEG could form PPR easily in water by host-guest interaction [26]. However, the hydrogen bonds between adjacent α-CDs on PPR would form hydrophobic crystalline microdomain [27], which decreased the water-solubility and increase the crystallinity of PPR. The addition of urea or modification of α-CDs with hydrophilic compounds could improve the architecture of PPR nanoparticles [28, 29], however, the modification was complicated and would increase the PPR cytotoxicity. Here we reported a simple way to modify PPR with mPEG-protoporphyrin IX (PpIX) as blocking agent. The PpIX was not only photochemically activated to achieve the therapeutic effect but also enhance drug loading via π-π interaction with anticancer drug doxorubicin (DOX) [1, 30]. The drug loaded PPR nanoparticle exerted synergistic PDT and chemotherapy (Fig. 1). The PPR was synthesized according as shown in Fig. S1 (Supporting information). The PPR NPs were prepared by the host-guest interaction between PEG and α-CDs. The structures and molecular weights of mPEG-NH2, mPEG-PpIX and the final PPR were characterized by 1H NMR, MS and XRD as presented in Figs. 2, S2 and S3 in Supporting information. In Fig. 2A, the characteristic peaks of -CH2 neighbored to -NH2 observed at δ 3.11 demonstrated the success of transition hydroxyl of mPEG to amine group of mPEG-NH2. The intensity calculation of peak 3 revealed that nearly all the hydroxyl groups in mPEG segments were transformed into amino groups. After introducing PpIX into mPEG-NH2 via coupling reaction, a characteristic peak (7) assigned to the methylene nearby amide bond was observed, indicating the success of coupling reaction. All signals (4-6) attributed to PpIX with the chemical shift d 10.2-10.42, 8.46-8.64, 6.20-6.30 and 6.40-6.60 as shown in Fig. 2B. Aside from the appearance of proton peaks of PpIX, the proton peaks at δ 3.51 (7) assigned to mPEG blocks were observed. Moreover, the mass spectra (Fig. S2) provided further evidences for the successful synthesis of mPEG-NH2 and mPEG-PpIX. To PPR (Fig. 1C), the characteristic peaks of α-CD (8-11) with the chemical shift at δ 4.80, 5.50, 5.43, 4.48 appeared. Meanwhile, the peaks of 4-6 at the chemical shifts δ 10.20-10.42, 8.46-8.64, 6.20-6.30 and 6.40-6.60 belonged to PpIX and the proton peak (7) attributed to PEG segment at the chemical shift δ 3.51 were all observed, the results meant α-CDs successfully parceled the mPEG segment via hostguest interaction. The formation of PPR NPs was further verified by XRD (Fig. S3), the diffraction peaks at 2θ = 19.3º and 23.4º belonged to mPEG-NH2 blocks were observed, while the peak 2θ = 23.4º disappeared and the peak 2θ = 19.3º was offset to 19.7º for PPR nanoparticles, which indicated that mPEG-PpIX amphiphilic polymer self-assembled with α-CDs and parceled the mPEG blocks, and further formed clathrate tunnel structure [31, 32].

|
Download:
|
| Fig. 1. The illustration of polypseudorotaxane NPs with combination of PDT and chemotherapy. | |
{kind=link}

|
Download:
|
| Fig. 2. The 1H NMR spectra of (A) mPEG-NH2 (CDCl3), (B) mPEG-PpIX (DMSO-d6), and (C) PPR (DMSO-d6); (D) DLS and AFM images of the blank NPs and DOX-loaded NPs; (E) The CMC of PPR. | |
{kind=link}
The blank PPR NPs and DOX-loaded PPR NPs were prepared, and the morphology and size distribution of these nanoparticles were determined by AFM and DLS (Fig. 2D). The NPs fabricated by PPR polymer in water were spherical in shape with average size of 41 nm and homogeneous distribution. The sized of DOX-loaded NPs increased to 89 nm for the DOX encapsulation, and the AFM spectrum showed the NPs were also spherical and homogeneous. These good properties ensured these NPs could reach the tumor microenvironment and penetrate the cell membrane easily [33]. Critical micelle concentration (CMC) for PPR polymer was also investigated to certify the formation of polymeric NPs and evaluate their stability. As presented in Fig. 2E, PPR exhibited a low critical value with CMC of 9.3 μg/mL, implying that the polymer can easily self-assemble into NPs and the formed nanoparticles were very stable even at high dilution. This property was beneficial for intravenous application. Moreover, the drug loaded efficiency of the PPR was calculated to be 9.93%, when thirteen α-CDs were in PPR (calculated by the 1H NMR, as shown in Fig. S4 in Supporting information).
Confocal laser scanning microscopy (CLSM) and flow cytometry tests were performed to study the internalization efficiency of DOX-loaded PPR NPs (Fig. 3). In CLSM, after two hours' incubation, weak fluorescence of PpIX (PpIX emitted red fluorescence in PPR group). No generation of ROS was found in blank and DOX loaded PPR groups. As the incubation time was extended to 5 h, strong green fluorescence of ROS for blank and DOX-loaded PPR groups was monitored, which suggested that more nanoparticles were internalized into the cells and strong ROS was generated under laser irradiation. The intensity of both red and green fluorescence increased with increasing incubation time. The internalization of NPs was further verified by flow cytometry as shown in Fig. 3, similar results were obtained and the results were consistent with CLSM data.

|
Download:
|
| Fig. 3. (A) The confocal laser scanning microscopy images of HepG2 cells with PPR blank NPs, DOX-loaded NPs, free DOX·HCl for 2 h and 5 h, fluorescence channel of ROS or DOX was carried out, the scale bar was 25 mm; flow cytometry histogram profiles of HepG2 cells incubated with free DOX·HCl and DOX-loaded NPs for 2 h (B) and 5 h (C), the DOX concentration was 7.5 μg/mL. | |
{kind=link}
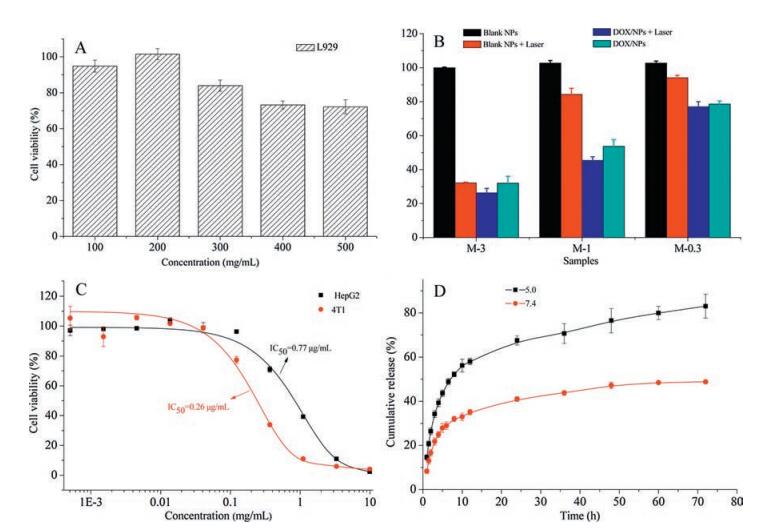
The cytotoxicity of PPR NPs was evaluated as shown in Fig. 4A. It was found that cell viability of NPs was as high as 80% even the concentration of the PPR nanoparticles as high as 300 μg/mL, revealing the good biocompatibility of the NPs.

|
Download:
|
| Fig. 4. (A) The cytotoxicity of PPR blank NPs incubated with L929 fibroblasts cells for 48 h. (B) Cell viability of HepG2 cells incubated with PPR blank NPs and DOX-loaded NPs without or with laser irradiation, three samples with different concentration of blank or drug-loaded micelles were used, M-0.3 (4.0 μg/mL), M-1 (12.1 μg/mL), M-3 (36.2 μg/mL). (C) The IC50 of DOX-loaded NPs against HepG2 and 4T1 without laser. (D) Drug release profiles of DOX-loaded NPs in different buffer solution (pH 5.0 and pH 7.4). The results were expressed as mean ± SD (n = 3). | |
{kind=link}
Laser irradiation was carried out to evaluate the in vitro anticancer activity (Fig. 4B). The concentrations of NPs were M-0.3 (4.0 μg/mL), M-1 (12.1 μg/mL) and M-3 (36.2 μg/mL). To blank PPR NPs without laser irradiation, no cytotoxicity was found. The cell viabilities of blank PPR NPs of M-0.3, M-1 and M-3 were 94.0%, 84.3%, and 37.2% with laser exposure, and the laser wavelength was 620-630 nm, generated by a diode laser. The cell viabilities of DOXloaded NPs with laser exposure were only 77.0% for M-0.3, 45.4% for M-1, and 26.3% for M-3. These results revealed the DOX loaded PPR NPs exposed in laser irradiation exhibited synergistic effect. The half maximal inhibitory concentration (IC50) of DOX loaded PPR NPs to HepG2 and 4T1 cells were 0.77μg/mL and 0.26 μg/mL, respectively (Fig. 4C). It revealed that the anticancer activity was different to different cell lines. The drug release behaviors of DOXloaded PPR NPs were investigated in buffer solution with pH 5.0 and pH 7.4 (Fig. 4D). A rapid initial burst release and a slow following release phase were observed in all the release profiles, the DOX release in pH 5.0 was higher relatively as DOX was protonated in acid medium.
In conclusion, a simple method of preparing PPR NPs was developed by self-assembled mPEG-PpIX and α-CDs via host-guest interaction for synergistic PDT and chemotherapy. The results demonstrated the DOX-loaded NPs exhibited good cellular uptake and low IC50. The DOX loaded PPR NPs showed excellent therapeutic efficacy for the combined PDT and chemotherapy.
AcknowledgmentsThe authors thank for the financial support of National Natural Science Foundation of China (No. 51573111), Program for Changjiang Scholars and Innovative Research Team in University (No. IRT-15R48).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.07.029.
| [1] |
X. Deng, Y. Liang, X. Peng, T. Su, S. Luo, Chem. Commun. 51(2015) 4271-4274. DOI:10.1039/C4CC10226F |
| [2] |
X. Yao, Z. Tian, J. Liu, Y. Zhu, N. Hanagata, Langmuir 33(2017) 591-599. DOI:10.1021/acs.langmuir.6b04189 |
| [3] |
Y. Liang, W. Gao, X. Peng, et al., Biomaterials 100(2016) 76-90. DOI:10.1016/j.biomaterials.2016.05.023 |
| [4] |
N. Li, T. Li, C. Liu, et al., J. Biomed. Nanotechnol. 12(2016) 878-893. DOI:10.1166/jbn.2016.2226 |
| [5] |
X. Wang, Y. Yang, Y. Zhuang, et al., Biomacromolecules 17(2016) 2920-2929. DOI:10.1021/acs.biomac.6b00744 |
| [6] |
J. Shen, L. Zhao, G. Han, Adv. Drug Deliv. Rev. 65(2013) 744-755. DOI:10.1016/j.addr.2012.05.007 |
| [7] |
T. Wang, D. Wang, H. Yu, et al., ACS Nano 10(2016) 3496-3508. DOI:10.1021/acsnano.5b07706 |
| [8] |
X. Yi, D. Zhao, Q. Zhang, et al., Nanotechnology 28(2017) 85603. DOI:10.1088/1361-6528/aa5715 |
| [9] |
G. Obaid, M. Broekgaarden, A. Bulin, et al., Nanoscale 8(2016) 11253-12471. |
| [10] |
L. Wang, T. Wu, D. Wu, Y. You, ACS Appl. Mater. Interfaces 8(2016) 19238-19244. DOI:10.1021/acsami.6b04327 |
| [11] |
P. Avci, S.S. Erdem, M.R. Hamblin, J. Biomed. Nanotechnol. 10(2014) 1937-1952. DOI:10.1166/jbn.2014.1953 |
| [12] |
M. Broekgaarden, R. Weijer, A.C. van Wijk, J. Biomed. Nanotechnol. 13(2017) 204-220. DOI:10.1166/jbn.2017.2327 |
| [13] |
C. He, D. Liu, W. Lin, ACS Nano 9(2015) 991-1003. DOI:10.1021/nn506963h |
| [14] |
Y. Li, T. Lin, Y. Luo, et al., Nat. Commun. 5(2014) 4712. DOI:10.1038/ncomms5712 |
| [15] |
I. Roy, T.Y. Ohulchanskyy, H.E. Pudavar, et al., J. Am. Chem. Soc. 125(2003) 7860-7865. DOI:10.1021/ja0343095 |
| [16] |
K. Ogawara, T. Shiraishi, T. Araki, et al., Eur. J. Pharm. Sci. 82(2016) 154-160. DOI:10.1016/j.ejps.2015.11.016 |
| [17] |
J. Han, W. Park, S. Park, K. Na, ACS Appl. Mater. Interfaces 8(2016) 7739-7747. DOI:10.1021/acsami.6b01664 |
| [18] |
G. Liu, J. Hu, G. Zhang, S. Liu, Bioconjug. Chem. 26(2015) 1328-1338. DOI:10.1021/bc500548r |
| [19] |
Y. Cakmak, E.C. Sundus, D.A. Leigh, J. Am. Chem. Soc. 138(2016) 1749-1751. DOI:10.1021/jacs.6b00303 |
| [20] |
Y. Li, Y. Chen, H. Dong, C. Dong, Med. Chem. Commun. 6(2015) 1874-1881. DOI:10.1039/C5MD00299K |
| [21] |
R. Chen, H. Jiang, H. Gu, et al., Org. Lett. 17(2015) 4160-4163. DOI:10.1021/acs.orglett.5b01910 |
| [22] |
F. Hosseini, A. Panahifar, M. Adeli, et al., Colloids Surf. B-Biointerfaces 103(2013) 652-657. DOI:10.1016/j.colsurfb.2012.10.035 |
| [23] |
F. Li, J. He, M. Zhang, K.C. Tam, P. Ni, RSC Adv. 5(2015) 54658-54666. DOI:10.1039/C5RA06156C |
| [24] |
A. Kulkarni, K. DeFrees, R.A. Schuldt, et al., Mol. Pharm. 10(2013) 1299-1305. DOI:10.1021/mp300449t |
| [25] |
L. Qin, D. Cao, H. Huang, J. Biomed. Nanotechnol. 12(2016) 261-273. DOI:10.1166/jbn.2016.2155 |
| [26] |
J. Chang, Y. Li, G. Wang, B. He, Z. Gu, Nanoscale 5(2013) 813-820. DOI:10.1039/C2NR32927A |
| [27] |
J. Li, A. Harada, M. Kamachi, Polym. J. 26(1994) 1019-1026. DOI:10.1295/polymj.26.1019 |
| [28] |
J. Cai, L. Zhang, C. Chang, et al., ChemPhysChem 8(2007) 1572-1579. DOI:10.1002/(ISSN)1439-7641 |
| [29] |
H. Dong, Y. Li, S. Cai, et al., Angew. Chem. Int. Ed. 47(2008) 5573-5576. DOI:10.1002/anie.v47:30 |
| [30] |
Y. Liang, X. Deng, L. Zhang, et al., Biomaterials 71(2015) 1-10. DOI:10.1016/j.biomaterials.2015.08.032 |
| [31] |
A. Harada, J. Li, M. Kamachi, Macromolecules 26(1993) 5698-5703. DOI:10.1021/ma00073a026 |
| [32] |
K. Olson, Y. Chen, G.L. Baker, J. Polymer Sci. Part A 39(2001) 2731-2739. DOI:10.1002/(ISSN)1099-0518 |
| [33] |
S.D. Perrault, C. Walkey, T. Jennings, H.C. Fischer, W.C.W. Chan, Nano Lett. 9(2009) 1909-1915. DOI:10.1021/nl900031y |