 2017, Vol. 28
2017, Vol. 28 



b Laboratory of Molecular and Translational Medicine, Key Laboratory of Birth Defects and Related Diseases of Women and Children of Ministry of Education at Sichuan University, West China Second University Hospital, Sichuan University, Chengdu 610061, China
Hydrogels are three-dimensional networks composed of hydrophilic polymer chains, which possess the ability to absorb a large amount of water (up to thousands of times their dry weight) to be plastic and mimic the in vivo circumstance [1]. Nowadays, the design strategies of the hydrogels have been extensively studied and hydrogels prepared via a diverse range of chemical and physical interactions often exhibit outstanding characteristics such as stimuli-responsiveness, biomimetic, biocompatibility, tunable and reversible physical/chemical properties [2-6]. Nevertheless, the structural and functional integrity of the hydrogels is jeopardized by either the external mechanical force or chemical erosion during the hydrogels application, especially in sophisticated in vivo environment [1, 7]. Hence, hydrogels designed with the intrinsic ability to self-repair when exposed to destructive factors served as an appealing candidate to address this issue.
Hydrogels with the ability to achieve autonomous repair to its initial set of properties in response to damage are defined as "selfhealing hydrogels" [8, 9]. Diverse self-healing hydrogels with unique properties have been developed. For instance, hydrogels able to self-heal at low pH were used as sealants for acid containing vessels and showed potential application in treatment of gastric ulcer and perforations [10]. Hydrogels with shear-thinning behavior could be injected trough a syringe and healed autonomously, and were thus exploited as self-supporting 3D printing inks [11]. Besides, injectable self-healing hydrogels could be facilely delivered in vivo without obvious impairment to the body, since no surgical incision was needed for hydrogels embedment [12]. Therefore, compared with conventional hydrogels, selfhealing hydrogels exhibit greater applied potential in different fields.
The process and outcome of self-healing are characterized mainly in two aspects: one is the restoration of micro-and macrostructure (morphology/topography) [13-15], and another is the recovery of mechanical and rheological properties [16-18]. Selfhealing mechanism lies in the dynamic equilibrium of interactions and requires a "mobile phase" to initiate and assist self-healing process [19]. Strategies to fabricate self-healing hydrogels can be divided into two categories: the non-covalent bonding (physical crosslinking) and the dynamic covalent bonding (chemical crosslinking). Non-covalent bonding includes hydrophobic bonding [20-22], hydrogen bonding [23, 24], host-guest interactions [25-27], ionic bonding [28, 29], etc. Dynamic covalent bonding includes imine bonds [30, 31], acylhydrazone bonds [32, 33], boronate ester bonds [34-36], disulfide bonds [37], Diels-Alder reactions [13], oxime bonds [38], etc. In both system, the continuous and dynamic dissociation and recombination of bonds confers the self-healing ability.
In this review, the current progress made in self-healing hydrogels was systematically discussed, including its design, mechanism and biomedical application. First, methods to qualitatively and quantitatively evaluate self-healing process and outcome were summarized. Then, we discussed about the designing strategies and self-healing mechanism of self-healing hydrogels. What's more, in vivo application of self-healing hydrogels was summarized. Future development of self-healing hydrogels was also illustrated.
2. Testing for self-healing hydrogelsDiverse methods have been developed to fully characterize the properties of hydrogels, covering from the crosslinking kinetics to biological applications [17, 39]. As hydrogels equipped with the unique property of self-healing, it is essential to fully value the selfhealing performance by observing the healing process and quantifying the healing outcome. Techniques for the evaluation of self-healing hydrogels can be classified into two categories. One is to observe and characterize the morphology or topography of self-healing hydrogel, including macroscopic visual observation [23, 39] and machine-aided optical microscopy [13, 14], scanning electron microscopy (SEM) [15, 40], Scanning electrochemical microscopy (SECM) [13] and atomic force microscopy (AFM) [14, 16]. Another is to quantify the mechanical properties, including general gravity/stretching/bending resistance test [23, 39], tensile strength test [18, 29], compressive strength test [17] and rheological test [19, 41]. Healing efficiency could be calculated by comparing the healed hydrogels with the pristine one via parameters affecting the healing behaviors such as crosslinking density, free monomers, healing time, temperature and other external stimulus [17, 29].
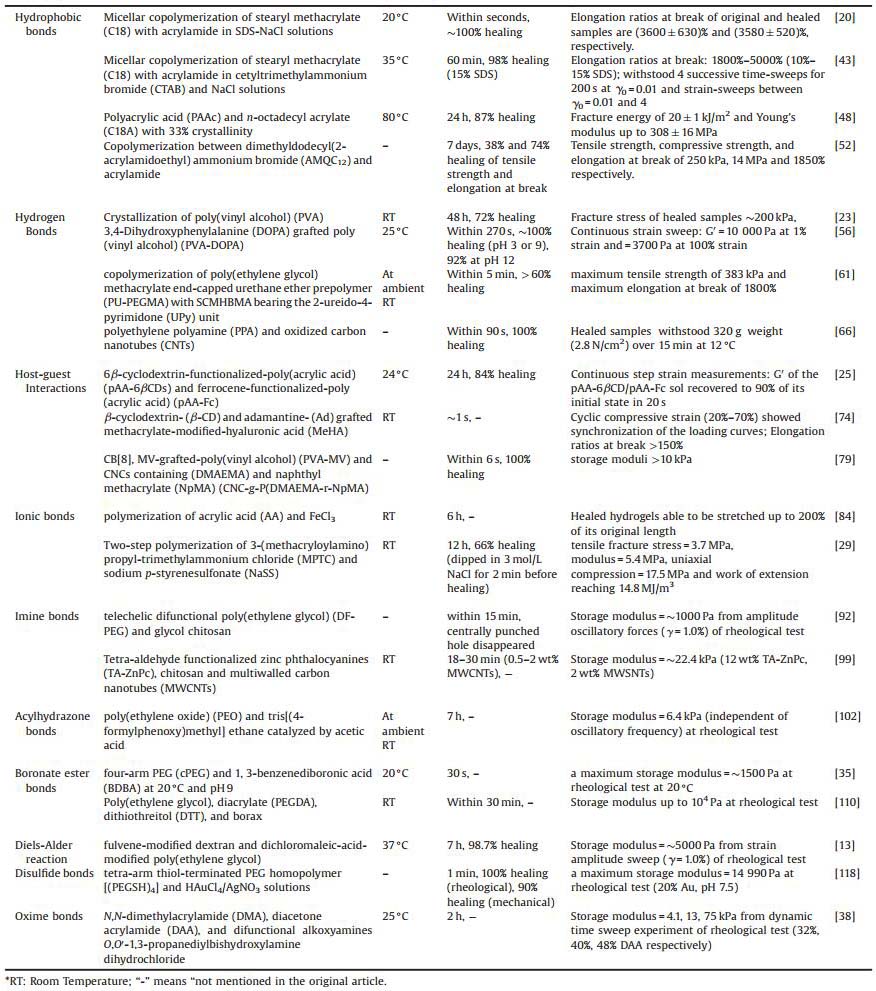
Characterization of morphology/topography of the hydrogels can be simply done by visual observation [23, 39]. Two cut pieces of hydrogels were put into contact without any external intervention. After a period of time (from seconds to hours), the fusion of cut pieces was viewed. The extent of fusion often indicates the magnitude of interaction between two cut pieces and mobility of the crosslinking network. Often, pigments like rhodamine B and typanblue were added into the hydrogels to allow distinguishable interfaces [23, 39] (Fig. 1a). However, details like the extent to which the hydrogels have healed, the longitudinal and horizontal depth/width of the healing interfaces and the restoration of the crosslinks can hardly be observed at macroscopic scale. SEM helps to reveal the morphology of the hydrogels at nanometer scale, illustrating crosslinking density and framework changes before and after healing [15, 40]. SECM provides spatial and temporal information about self-healing procedure. A three-dimensional illustration of the scratch areas on hydrogels is made and healing efficiency could be calculated by comparing the scratch depth and width at different time [13]. AFM is also available for morphology/ topography characterization. For example, Wang and coworkers employed AFM to monitor the whole healing process, and testified the importance of "mobile phase" in the generation of self-healing [14, 16] (Fig. 1b). Importantly, morphology/topography characterization of self-healing hydrogels helps with the continuous observation of healing process and qualitative evaluation of outcome. To further quantify the healing efficiency and healing ratio, mechanical characterization related to parameters like Young's modulus and viscoelastic properties should be performed.

|
Download:
|
| Fig. 1. Testing methods for self-healing hydrogels. (a) Visual evaluation of self-healing behaviors of PVA-based hydrogels. Two cut pieces of hydrogels were brought into contact for 12 h at room temperature without any other interventions and the healed one was then tested to resist bending (lower left) and stretching (about 100% extension) (lower right). Reproduced with permission [23]. Copyright 2012, American Chemical Society (b) Atomic force microscope (AFM) showed a microscopic dimension of the healing process of scratched disulfide bonding-based hydrogels. Cut width healed from 7.8 mm (left) to 4.2 μm (middle) after 12 h by sprinkling a trace amount of water and almost 0 μm (right) by repeating the process after 12 h. Reproduced with permission [14]. Copyright 2016, The Royal Society of Chemistry. (c) Alternate step-strain experiment showed the change in storage moduli (G′) and loss moduli (G″) of Dex-l-PEG hydrogels. Reproduced with permission [13]. Copyright 2013, Wiley-VCH. | |
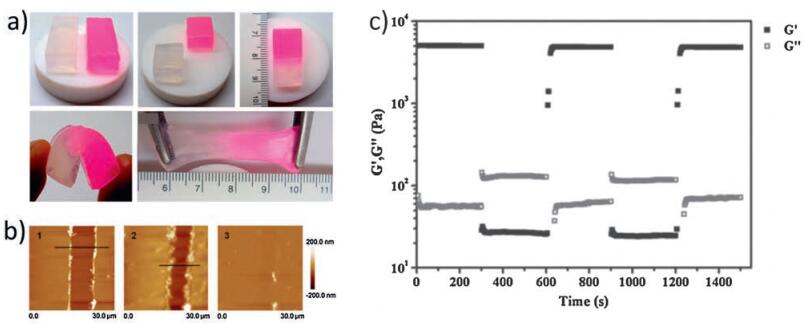
Mechanical characterization of the self-healing hydrogels are routinely carried out. One approach is the gravity/stretching/ bending resistance test to evaluate the ability of hydrogels to maintain the healed state [23, 39]. The healed hydrogel was lifted by tweezers to test the ability to withstand its own weight [23]. Sometimes, stretching or bending were further applied to demonstrate the superior healing ability [39]. Tensile test, usually uniaxial elongation measurement, is performed on hydrogels at large deformation. Stretch rates in tensile test are tunable and stress-strain curves can be plotted. Fracture stress and Young's modulus (E) are recorded as important parameters evaluating the properties of the hydrogels. Elongation at break (elongation ratio) is most frequently used to determine the healing efficiency [18, 39]. However, for hydrogels which are too soft, flexible and fragile, tensile test is inoperable because hydrogels are either too dynamic to shape into required shape or unable to bear clamping. Alternatively, compressive test is applied, in which a wedgeshaped plunger is often used to determine the strength on joint interfaces of healed hydrogels [25, 26]. Rheological test focuses on the viscoelastic properties of the hydrogels [13, 19, 41]. Continuous step-strain measurements are frequently performed to evaluate the self-healing and reversibility of the hydrogels (Fig. 1c). When hydrogels are exposed to a large strain (e.g., γ = 1000%), the storage modulus (G′) dropped sharply ( < loss modulus, G″), indicating the collapse of crosslinking in the hydrogels (sol state). Once the large strain is replaced by a small strain (e.g., γ = 1%), the G0 recovered in a step-wise manner. When G′ becomes larger than G″, it means the dynamic crosslinking transforms from sol to gel state. The time it takes to fully recover, the recovered storage modulus compared to the original one, and the numbers of cycles hydrogels could bear reveal the self-healing properties of the hydrogels [13, 19, 41].
3. Self-healing mechanismSelf-healing hydrogels should be designed according to its intended application. Though the design strategies and mechanism of self-healing hydrogels varies, the general principles should be followed: (1) materials and reactions should be nontoxic and economical; (2) hydrogels can heal autonomously, sometimes in a stimuli-responsive way; (3) healing occurs in a repeatable and efficient way; (4) healing occurs both morphologically (micro-and macro-scale) and mechanically (mechanical and rheological properties); (5) a complete retention of morphology and mechanical properties after healing would be preferable. Based on those principles, the self-healing mechanism can be generally divided into two categories: the non-covalent bonding (physical crosslinking) and dynamic covalent bonding/chemistry (chemical crosslinking).
Non-covalent bonding mainly includes hydrophobic bonding [20-22], hydrogen bonding [23, 24], host-guest interactions [25-27], and ionic bonding [28, 29]. Non-covalent bonding is thought to be mechanically weak compared with covalent bonding due to its weak intermolecular force and reversible interactions. In recent years, "tough" self-healing hydrogels have been successfully fabricated by incorporating nano-composites and interpenetrating polymer network into the system (reviewed in Ref. [8]). Dynamic covalent bonding/chemistry, on the other hand, adopts the idea of rapid equilibrium, which means the reversible assemble and disassemble of components occur simultaneously and continuously in the whole reaction system. Hydrogels based on dynamic covalent chemistry are entailed with the ability of self-healing because the dynamic covalent bond crosslinking the networks could break and reform autonomously. Besides, hydrogels prepared from dynamic covalent bonding could exhibited some unique properties like pH-, thermo-and redox-responsiveness due to the nature of covalent bonds. Strategies used in dynamic covalent chemistry are diverse. In this review, we focused on the main types of hydrogels, which formed by imine bonds [30, 31], acylhydrazone bonds [32, 33], boronate ester bonds [34-36], disulfide bonds [37], Diels-Alder reactions [13] and oxime bonds [38]. Schematic illustration of the self-healing mechanism was shown in Fig. 2. Examples of self-healing hydrogels regarding the healing mechanism, materials, healing conditions, healing efficiency and mechanical properties were summarized in Table 1.

|
Download:
|
| Fig. 2. Schematics of the self-healing mechanism. | |
|
|
Table 1 Examples of self-healing hydrogels (Healing mechanism, materials, conditions, efficiency and mechanical properties). |
{kind=link}
{kind=link}
3.1. Noncovalent bonds (physical bonds) 3.1.1. Hydrophobic bonds
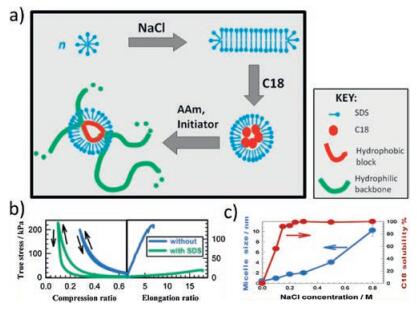
Hydrophobic bonds are reversible noncovalent interaction existing among non-polar hydrophobic groups. Polymer chains with hydrophobic monomers crosslink to form a network which could easily regain their structure via hydrophobic bonds when disturbed [42]. Methods hiring hydrophobic bonds to the formation of self-healing hydrogels have been developed, among which the most extensively studied technique is called "micellar polymerization" [16, 20-22, 43-52] (Fig. 3a). Successful micellar polymerization requires four crucial components in the reacting system: hydrophobic units, hydrophilic units, surfactants and electrolytes. Hydrophobic units solubilizing in surfactants-and electrolytes-containing aqueous solution could copolymerize with hydrophilic units to form self-healing hydrogels [16, 20-22]. For example, Okay and colleagues developed a hydrogel based on copolymerization of large hydrophobic monomer stearyl methacrylate (C18) with hydrophilic monomer acrylamide (AAm) in aqueous solution containing surfactant sodium dodecyl sulfate (SDS) and electrolyte NaCl [16, 20-22]. Strong hydrophobic association among C18 units endowed the hydrogels with excellent mechanical properties with elongation ratio up to 3600%. Self-healing experiment showed 100% recovery of the hydrogels within seconds at room temperature [16]. SDS greatly influenced mechanical property and self-healing behavior of the self-healing hydrogels [21] (Fig. 3b). In the reaction system, SDS formed wormlike micelles encompassing C18 monomers, which induced local solubilization of the hydrophobic association and thus the hydrophobic interaction among the polymer chains is reversible. Extraction of SDS from the hydrogels resulted in the loss of self-healing ability due to irreversible crosslinks and the increased mechanical strength because of strong hydrophobic associations [21]. Addition of electrolytes such as NaCl into solutions could weaken the electrostatic interaction, which helped with the solubilization of hydrophobes and micellar growth [20] (Fig. 3c). Difference in the lengths and concentrations of alkyl side chains also affected the self-healing ability of the hydrogels [22]. Increasing alkyl side chain length from C16 to C18 resulted in improved healing efficiency from 29% to 34% at room temperature. Replacing acrylates with methacrylate leaded to an increase of healing efficiency from 34% to 88% after 30 min. This is because the incorporation of methacrylate backbone hindered the alignment of alkyl chains, which limited the flexibility of the backbone chains and subsequently increased the portion of free hydrophobic blocks available for self-healing [22]. With the assistance of cryo-electron miscrosocpy (cryo-EM) and small-angle neutron scattering (SANS) techniques, the authors exploited the nanoscale structural and dynamical change of the hydrogels, illuminating the underlying mechanism relating to self-healing [16]. Introduction of hydrophobic and hydrophilic monomers into SDS solutions led to conformational transition of wormlike SDS micelles from crosssectional radius of 1.6 nm to spherical micelles with radius of 2.4 nm. Intralayer mobility of the micelles initiated the self-healing process. Circular shape formed at the cutting surface and then complete healing occurred due to interlayer mobility [16].

|
Download:
|
| Fig. 3. Hydrophobic interaction-based self-healing hydrogels. (a) Illustration of self-healing hydrogels based on copolymerization of hydrophobic monomer C18 with hydrophilic monomer acrylamide (AAm) in aqueous solution containing surfactant SDS and electrolyte NaCl. (b) Tensile and compressive strength test of hydrogels with/without SDS. Reproduced with permission [21]. Copyright 2012, American Chemical Society. (c) NaCl helped with the solubilization of C18 and micellar growth. Reproduced with permission [20]. Copyright 2011, American Chemical Society. | |
{kind=link}
Mixing oppositely charged cationic surfactant cetyltrimethylammonium bromide (CTAB) with anionic charged surfactant SDS could also promote micelles growth resembling SDS-NaCl micellar system [44, 45]. The hydrogels demonstrated 98% healing efficiency after 60 min and 1800%-5000% elongation ratios at break. Increasing the size of micelles led to elevated degree of temporary spatial inhomogeneity and lifetime of hydrophobic associations, but decreased elongation ratio at break. Compared with SDS-NaCl system, CTAB-SDS hydrogels were more elastic because of a relatively longer alkyl chain length of CTAB and a consequent improved hydrophobic interactions [44].
Different hydrophilic monomers gave rise to distinct mechanical and self-healing properties [44-46]. Micellar copolymerization of acrylic acid with C18 in SDS-NaCl solution generated physical gels with improved mechanical and self-healing properties [44-46]. The polyacrylic acid (PAAc) hydrogel exhibited time-dependent dynamic moduli, 6-53 kPa Young's modulus, 41-173 kPa fracture stress and 1800%-5000% elongation ratios at break. Fractured hydrogels could heal autonomously and 60% healing efficiency was achieved after 30 min at room temperature. Hydrogels healing at 80 ℃ reached at least 80% healing efficiency. The stronger self-healing ability of PAAc gels compared with polyacrylamide (PAAm) gels was attributed to the aid of hydrogen bonding between carboxyl groups of PAAc chains [45]. Furthermore, copolymerizing acrylic acid with C12 in CATB solution produced hydrogels with shape memory behavior, high tensile strength (0.7-1.7 MPa) and elongation ratios at break (800%-900%) [44]. Similarly, replacing PAAm with poly(N, N-dimethylacrylamide) (PDMA) in SDS-NaCl solution formed a hydrogel which withstood 100% compressive strain and reached 4200% elongation ratio [46]. Besides aforementioned hydrophilic monomers, variance of hydrophobic monomers in the reaction system also affected the mechanical properties of hydrogels [47]. Fatty alcohol polyoxyethylene acrylate (AEO-AC) was used as hydrophobe to prepare hydrogels which possessed up to 318 kPa mechanical strength and 1000%-3000% elongation ratios. Varying the length and branches of the backbone let to different strength of hydrophobic association [44-46]. The introduction of crystalline domains along the alkyl side chains (PDMA or PAAc) greatly enhanced the mechanical strength of the hydrogels [48, 49]. For example, PAAc/C18A (n-octadecyl acrylate) hydrogels with 33% degree of crystallinity exhibited modulus E up to 308 MPa. Tensile fracture strength of 4-7 MPa could be obtained once the degree of crystallinity exceeded 10%. Healing efficiency was around 87% and the healed hydrogels withstood 138 ± 10 MPa compressive stress [48]. Applying heat above melting temperature induced the melt of crystalline domains of alkyl side chains and a subsequent liquefaction of the hydrogels. Therefore, hydrogels can be molded into any desired shape by heating [49].
A hybrid hydrogel combining physical and covalent network was synthesized via micellar copolymerization of acrylamide with C18 as physical cross-linker and N, N'-methylenebis(acrylamide) (BAAm) as chemical cross-linker [50]. Mechanical strength of the hydrogels increased with BAAm content. Self-healing was observed only if the cross-linker ratio X < 0.01 (molar ratio of BAAm to the monomer). Increasing X more than 0.01 led to the loss of self-healing ability due to restricted mobility of C18 blocks in inhomogeneous network [50]. Pixin et al. developed a surfactant-free micellar copolymerization system by introducing surfactant monomers (surfmers) with both surface activity and polymerizability. Surfmers formed stable micelles in aqueous solution without surfactants and could be either anionic (9 or 10-acrylamidostearic acid, NaAAS) [51] or cationic (dimethyldodecyl(2-acrylamidoethyl)ammonium bromide, AMQC12) [52]. For instance, hydrogels were prepared via copolymerization between NaAAS and acrylamide [51]. The hydrogels with 20% surfmer exhibited compressive strength up to 22.50 MPa at strain of 90% and 13-fold elongation length. Self-healing and pH-responsive behavior were also observed.
3.1.2. Hydrogen bondsHydrogen bonds (H-bonding) occur between two polar groups, namely hydrogen atom and highly electronegative atom such as nitrogen (N), oxygen (O) and fluorine (F). Though it is relatively weak compared with covalent or ionic bond, concerted H-bonding interactions contribute to the improved bond strength and thus hydrogels formation. For instance, poly(vinyl alcohol) (PVA) contains numerous hydroxyl moieties which could form intermolecular H-bonding [23, 53-55]. Applying freezing and thawing on PVA resulted in the formation of crystallites that cross-linked with each other by H-bonding and formed the network of the selfhealing hydrogels [23]. Hydrogels healing for 10 s could bear stress up to 10 kPa. After 48 h healing, hydrogels could recover to 72% of the initial tensile strength. However, the healing behavior of hydrogels was time-dependent. Prolonged separation time resulted in the loss of self-healing ability due to the formation of inter-or intra-chain H-bonding, which reduces the free hydroxyl groups available on cut surfaces [23]. Based on the same freezing/ thawing method, a melamine enhanced PVA self-healing hydrogel was reported [53]. Melamine formed multiple H-bonds with PVA, increasing mechanical property of the hydrogel. For example, the elongation at break and tensile strength increased 160% and 420% respectively compared with PVA hydrogels. Applying ultrasound to the hydrogels triggered shape recovery of the hydrogels by melting crystalline micro-domains (Fig. 4a). This responsiveness, added with its biocompatibility, showed potential applications in biomedical fields. Introducing physically cross-linked PVA into chemically cross-linked poly(ethylene glycol) (PEG) network lead to the formation of a double-network hydrogel [54]. Using this method, a chemically cross-linked hydrogel also exhibited selfhealing ability and shape memory property. Le et al. reported a poly (vinyl alcohol)-sodium alginate-borax (PVA-SA-borax) hydrogel based on hydrogen bonding [55]. The PVA-SA-borax hydrogels showed complete self-healing behavior in 3 h and demonstrated good barrier properties to hazardous chemicals like sodium cyanide, dichlorvos and phorate.

|
Download:
|
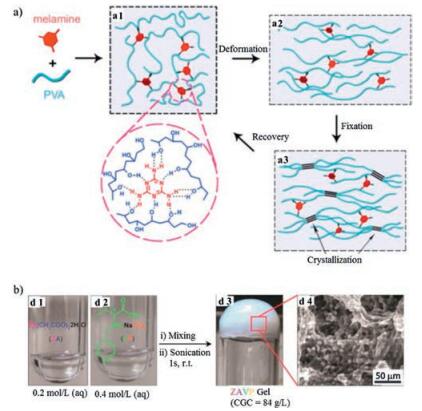
| Fig. 4. Hydrogen bonding-based self-healing hydrogels. (a) Schematic illustration of an ultrasound-triggered shape memory behavior of melamine enhanced poly (vinyl alcohol) (PVA) self-healing hydrogel. Hydrogen bonds were formed between melamine and PVA chains (a1). The hydrogels deformed (a2) and subsequently fixed by freezing/thawing under strain (a3) recovered its original shape when exposed to ultrasound treatment (a1). Reproduced with permission [53]. Copyright 2015, American Chemical Society. (b) Photographs of a metallo-hydrogel (d3) formed by mixing Zn(Ⅱ)-based salts (ZA) (d1) with l-valine-based ligand (VP) (d2). SEM-images of ZAPV hydrogel (d4). Reproduced with permission [68]. Copyright 2014, The Royal Society of Chemistry. | |
{kind=link}
3, 4-Dihydroxyphenyl-L-alanine (DOPA), a synthetic amino acid inspired from mussel foot protein 3 (Mfp3), contains catechol groups which were able to form H-bonding. Interactions among catechol groups were shown to play a critical role in initiating and accelerating self-healing process [24, 56, 57]. A DOPA-grafted-PVA self-healing hydrogel (PVA-DOPA) was prepared in metal-free aqueous solution [56]. Continuous strain sweep test demonstrated rapid self-healing property. G′ returned to initial value at 1% strain within 270 s after being been placed under 100% strain. Increasing pH from acid to basic resulted in higher dynamic moduli, which revealed the pH dependent behavior of the hydrogels. Lin et al. reported an ABA tri-block hydrogel with catechol-functionalized poly(N-isopropylacrylamide) (PNIPAM) as A block and poly(ethylene oxide) (PEO) as B block [57]. The hydrogels exhibited thermosensitivity with sol-to-gel transition when temperature increased from 12 ℃ to 37 ℃. The self-healing experiment demonstrated instant healing of the hydrogels when brought into contact and the healed hydrogels withstood vigorous shaking. Anti-biofouling property and shear-thinning behavior which enabled injectability were also shown.
Besides hydroxyl-based H-bonding (O--H…O), C=O and N--H could also interact to form H-bonds (N--H…O). For example, 2-ureido-4-pyrimidone (UPy) can form self-complementary quadruple H-bonds via dimerization, forming an AADD-DDAA array (A as acceptor, D as donor), thus could be used as the functional parts of the self-healing hydrogels [58]. Self-healing hydrogels adopting UPy units were first reported by Jiaxi and colleagues [59]. They copolymerized 2-(dimethylamino)-ethyl methacrylate (DMAEMA) with 2-(3-(6-methyl-4-oxo-1, 4-dihydropyrimidin-2-yl)ureido) ethyl methacrylate (SCMHBMA) to form a self-healing hydrogel. Rapid self-healing (within 5 min) occurred at pH from 7 to 8 at room temperature. Grafting UPy units onto alkyl chains/substituents (for example, adamantyl) could shield H-bonds from disruptive effect of water molecules by creating hydrophobic pockets [58]. Upy-modified-acrylamide [60] or -polyurethane [61] were used to fabricate self-healing hydrogels and thus obtained hydrogels were endowed with unique properties. For instance, the Upy-based polyurethane hydrogels exhibited excellent self-healing ability (60% recovery within 5 min), elasticity, robustness and toughness (maximum tensile strength: 383 kPa; maximum elongation at break: 1800%) [61]. Furthermore, a pHswitchable hydrogel was developed by bonding UPy units onto poly(ethylene glycol) (PEG) chains [62]. The delivery of Upy-PEGhydrogels containing HGF/IGF-1 into myocardial infarction via a 1-m long catheter to repair tissue defects was achieved due to the sol-gel switching of the hydrogels responding to pH change [62]. N--H…O hydrogen bonds are also known to contribute to the formation and maintenance the secondary structure of proteins [63]. Nicholas et al. reported an α-helical-peptide-based selfhealing hydrogel. H-bonding played a critical role in self-assembly of the 21-residue peptide into three-dimensional structure [63]. To enhance the mechanical strength of the hydrogels, N-acryloyl glycinamide (NAGA) containing dual amide motifs was utilized as the building block of the supramolecular self-healing hydrogels (PNAGA) [64]. The PNAGA hydrogels exhibited unique mechanical properties, with maximum tensile strength reaching 1.1 MPa, over 1400% elongation at break, 150 kPa Young's modulus and no swelling in water for months. The hydrogels also demonstrated pH stability and 84% healing efficiency after healing at 90 ℃ for 3 h. A self-healing hydrogel crosslinked by a DNA grafted polypeptide and an "X"-shaped DNA linker was developed [65]. The precise recognition through complementary interaction between single stranded DNA (ssDNA) via H-bonding endowed the hydrogel with designable responsiveness, rapid self-healing ability (complete healing within 5 min) and injectability. Furthermore, unlike polymeric self-healing hydrogels, this hydrogel was based on low molecular weight compounds, so an excellent thixotropic property was demonstrated.
As is mentioned, H-bonds are relatively weak and susceptible to external stimuli. Thus, strategies were developed to reinforce the bonding strength in network, one of which was the introduction of nano components [66, 67]. Simply mixing polyethylene polyamine (PPA) with oxidized carbon nanotubes (CNTs) led to the formation of a mechanically strong self-healing (PPA/CNT) hydrogel [66]. Apart from the weak H-bonding (N--H…H) exhibiting among PAA, CNTs formed strong H-bonding (N--H…O) with PPA, resulting in the multi-responsiveness (thermal, pH and NIR light) and increased mechanical behavior of the hydrogels. Hydrogels adhered on Teflon surface could withstand 320 g weight (2.8 N/cm2) over 15 min at 12 ℃. E12 ℃. Even dried PPA/CNT hydrogels could maintain self-healing property [66]. Exfoliated montmorillonite (MMT) clay nanoplatelet was another type of nano component utilized in the fabrication of hydrogels. Methylguanidine hydrochloride-grafted-xylose was copolymerized with MMT/PAAS suspension to form MMT/xylose self-healinghydrogels[67].Cuthydrogels could merge into one piece within minutes and withstand its own weight. MMT/xylose hydrogels demonstrated excellent thermal stability, with 90% residue weight of MMT remained under 600 ℃. In situ polymerization of acrylamide with MMT nanosheets also formed a hydrogel exhibiting exceptional mechanical properties, with fracture elongation reaching 11800% and up to 10.1 MJ/m3 fracture toughness [66].
Unique design of H-bonding-based self-healing hydrogels could also be seen in metallo-hydrogels [68, 69]. An amino acid-based, low-molecular-weight metallo-hydrogel was prepared by mixing a L-valine-based ligand with Zn(Ⅱ)-based salts [68] (Fig. 4b). Zn(Ⅱ)-complex underwent self-assembly via H-bonding to form a hydrogel. The hydrogel was responsive to thermal, mechanical, pH and chemical stimuli. Continuous step-strain measurements revealed the hydrogels could recover to its original state within 3 min (0.1% and 1000% cyclic strain). The hydrogels also exhibited high load-bearing ability, withstanding 60-fold of its own weight [68]. Besides, a rapid thixotropic response to mechanical stimuli was observed by large-amplitude oscillatory test and shakingresting cycle test. Noticeably, metal ions were highly selective in metallo-hydrogels. Only the presence of Zn(Ⅱ) salts led to hydrogel formation. Similarly, a tyrosine and Ni2+ based self-healable metallo-hydrogel was reported [69]. Grafting the hydrogel network with dangling hydrocarbon side chains (poly(acryloyl-6-aminocaproic acid)) (A6ACA) to mediate H-bonding-based selfhealing was reported by Ameya et al. [10]. The optimal balance of hydrophobic and hydrophilic groups on side chains enabled the sufficient interfacial interaction while minimizing the steric hindrance of the hydrogels. A6ACA hydrogels was pH-dependent and could achieve rapid self-healing within 2 s at pH ≤ 3.
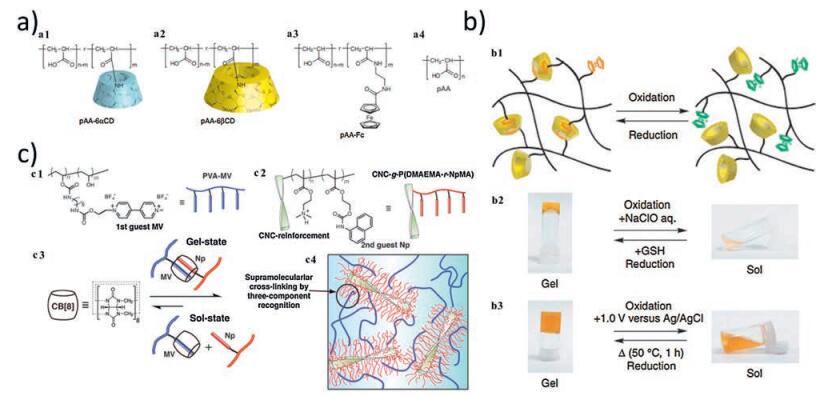
3.1.3. Host-guest interactionsHost-guest interaction is another type of non-covalent bond achieved by mutual molecular recognition of "host" and "guest" moieties. Generally, "host" moieties are macrocyclic molecules which could selectively accommodate and form inclusion complex with "guest" moieties through π-π stacking and hydrophobic interactions [70]. For instance, cyclodextrins (CDs), as soluble cyclic oligosaccharides with α-1, 4-glucose linked units, are composed of a hydrophobic internal cavity and could be designed as host moieties, encapsulating variable guest molecules [25, 26, 39, 70, 72, 76-78]. Masaki et al. synthesized a redox-responsive hydrogel based on 6β-cyclodextrin-functionalized-poly(acrylic acid) (pAA-6βCDs) and ferrocene-functionalized-poly(acrylic acid) (pAA-Fc) [25] (Fig. 5a). The self-healing behavior was achieved through reversible host-guest interaction between βCD and Fc, and the adhesion strength recovered to 84% of the initial state after 24 h. Rheological step-strain test revealed that pAA-6βCD/pAA-Fc hydrogel recovered to 90% of its initial G′ value within 20 s at 0.1% strain after the polymeric network collapsed at 200% strain. Redox treatment rendered hydrogel sol-gel transition by converting the reduced or oxidized state of Fc group, thus increased or reduced its affinity to βCD group respectively [25] (Fig. 5b). A supramolecular self-healing hydrogel crosslinked by poly(N, N-dimethylacrylamide-r-glycidol methacrylate-β-CD) [P(DMA-rGMA-CD)] and poly (N, N-dimethylacrylamide-r-2-hydroxyethylmethacrylate-Fc) [P(DMA-r-HEMA-Fc)] at room temperature and neutral pH exhibited electrochemical stimuliresponsive behavior [71]. Treating the hydrogel with oppositely charged potential (-0.10 V and +0.50 V) resulted in the sol-gel switching. Compared with redox reagents, electrochemical stimuli showed little adverse effect on the storage moduli of the hydrogel during cyclic responsive test, suggesting a facile way to control the reversible reaction of the inclusion complex [71]. Besides ferrocene (Fc), adamantine (Ad) is another type of molecules which could form inclusion complex (Keq ≈ 105 L/mol) with b-cyclodextrin (CD). Christopher and colleagues respectively grafted β-cyclodextrin (β-CD) and adamantine (Ad) onto hyaluronic acid (HA) and a supramolecular HA-βCD-Ad hydrogel instantly formed at room temperature upon mixing the two ingredients [11]. The hydrogel exhibited a shear-thinning and rapid self-healing behavior, thus was utilized as both "inks" and "support matrix" of 3D printing. The hydrogel could deform when squeezed out of the syringe and rapidly regain its printed structure on the substrate, enabling patterned design (for example, spiraling structure and cell pockets), which is impossible with conventional 3D printing method [11]. Proteolytic degradation of the hydrogel could be attained by binding β-cyclodextrin (β-CD) and adamantine (Ad) to hyaluronic acid (HA) via a peptide, the amino acid residues of which could be further tailored to control the degradation kinetics [72]. To strengthen the toughness and elastic moduli of hydrogels, methacrylate-modified-hyaluronic acid (MeHA) was designed as the polymer backbone of the bCD-Ad inclusion complex [73]. Therefore, a dual-cross-linking hydrogel (bCD-MeHA/Ad-MeHA) evolved, with host-guest assemblies (bCD-Ad) as the first network and photopolymerization-cross-linked methacrylate groups (MeHA) as the second network. Self-healing was achieved near-instantaneously (~1 s) and cyclic compressive stress-strain test showed little moduli loss between loading cycles (5 cycles at 0.5 N/min) [73]. βCD-MeHA/Ad-MeHA hydrogels also displayed a shear-thinning property and could be employed as 3D printing "ink" for "lay-by-lay" fabrication (>16 layers) [74]. To better address hydrogels' multi-functionalities in response to stimuli, Kohei et al. introduced a dual host-guest system by incorporating β-cyclodextrin-adamantine (βCD-Ad) and β-cyclodextrin-ferrocene (βCD-Fc) into one supramolecular network to form a βCD-AdFc self-healing hydrogel [75]. The self-healing was readily achieved within 2 h and the adhesive strength reached 68% of its initial state after 72 h. Applying redox reagents to hydrogel changed crosslinking degree by affecting the interaction between β-CD and Fc, which then caused the expansion-contraction and shape-memory behavior of the hydrogel [75]. Apart from ferrocene and adamantine, cholic acid could also form reversible host-guest complex with β -cyclodextrin [76]. Self-healing behavior was observed within less than 1 min and showed cross-section-size dependent property. Continuous step strain measurements (cycles of 10% strain then 1000% strain) revealed that hydrogel recovered to 97% of its initial storage moduli within 30 s. When the shear rate increased from 0.01 to 100 s-1, the hydrogel exhibited a transition from shear-thickening to shear-thinning behavior [76]. Polypeptide hydrogels based on cholesterol (Chol)-modified triblock poly (L-glutamic acid)-block-poly(ethylene glycol)-block-poly(L-glutamic acid) ((PLGA-b-PEG-b-PLGA)-g-Chol) and β-cyclodextrin (β-CD)-modified poly(L-glutamic acid) (PLGA-g-β-CD) were reported [39]. Self-healing was accomplished within 1 min and alternate step strain test (1% strain and then 200% strain) revealed full recovery of storage and loss moduli. Its ability to construct stable multilayer structure, added with good degradability and biocompatibility, indicated promising biomedical application of the hydrogel [39]. Pluoric F108 (PEG-b-PPG-b-PEG) exhibited high affinity to βCD, so an alginate-graft-β-CD/Pluoric F 108 supramolecular hydrogel was synthesized, which showed thermo-responsiveness and eminent self-healing property. Rheological stepstrain test revealed that the hydrogel recovered to its original mechanical properties at 0.5% strain within 10 s after being placed under 250% strain [77]. Hui et al. described a room-temperature phosphorescence (RTP) self-healing hydrogel based on interaction between α-bromonaphthalene (α-BrNp) polymer (poly-BrNp) and β -cyclodextrin (β-CD) polymer (poly-β-CD). The RTP property was attributed to the host-guest inclusion complex [78].

|
Download:
|
| Fig. 5. Host-guest interaction-based self-healing hydrogels. (a) Chemical structures of the host polymer pAA-6αCD (a1) and pAA-6βCD (a2); the guest polymer pAA-Fc (a3); the side chains pAA (a4). (b) Illustration of the sol-gel transition behavior of pAA-6βCD/pAA-Fc hydrogels (b1). Adding chemical reagents NaClO aq. (oxidant) to the hydrogel induced the gel to sol transition, and subsequent GSH (reductant) reversed the sol state to gel state (b2). Electrochemical oxidation and reduction also induced the sol-gel switching (b3). Reproduced with permission [25]. Copyright 2011, Macmillan Publishers Limited. (c) Schematic illustration of hydrogels based on nanocrystals (CNC) and CB [8] host-guest interaction. Three-component host-guest recognition occurred among first-guest PVA-MV (c1), second-guest CNC-g-P(DMAEMA-r-NpMA) (c2) and host motif CB[8] (c3). The crosslinking network of dynamic hydrogels are composed of the "soft" polymer chains (PVA-MV) bridging the "hard" brush-like CNC domains (c4). Reproduced with permission [79]. Copyright 2014, WILEY-VCH. | |
{kind=link}
Besides guest moieties, host moieties also influence the mechanics and self-healing of the hydrogels. Takahiro and colleagues reported β-cyclodextrin-adamantine (βCD-Ad) and α-cyclodextrin-n-butyl (aCD-nBu) hydrogels, which displayed different self-healing ability. βCD-Ad hydrogels achieved almost full recovery of the original adhesion strength after 24 h while only 74% restoration was reached in αCD-nBu hydrogels [26]. Cucurbit [n]uril (n = 5-8, 10) (CB[n]s) are another family of macrocyclic host moieties, among which CB[8] owns a relatively large cavity volume and is able to accommodate two guests through π-π stacking [27, 79, 80] (Fig. 5c). A cellulosic-derivatives-based ultrahigh-watercontent supramolecular hydrogel was formed through ternary interaction among CB[8] (host), MV-grafted-poly(vinyl alcohol) (PVA-MV) (guest) and naphthyl-functionalized cellulose (HEC--Np) (guest) [27]. Up to 99.7% content of the hydrogel is comprised of water, yet it still showed highly elastic property (tanδ = G′/G″ ≤ 0.25). Step-rate time-sweep measurements revealed rapid-recovery of the hydrogels, which also showed responsiveness to multi-stimuli including redox reagents and competing guests. Based on the same reversible host-guest complex, a cellulose nanocrystals (CNC)-based hydrogel was prepared [80]. The "soft" polymer chains and "hard" brush-like CNC domains, combined with CB[8]-MV-Np complex, provide the hydrogel with high storage moduli (>10 kPa), fast sol-gel switching ( < 6s) and rapid self-healing properties [79]. Self-healing hydrogels with the aromatic amino acids as guest molecules and cucurbit[8]uril (CB [8]) as host molecules were synthesized. Phenylalanine and Tryptophan could respectively formed 2:1 homo-ternary complexes with CB[8]. The nontoxic guest moieties in the hydrogel indicate good biocompatibility and could be further introduced into biomedical application [80].
The selection of backbone chains guest and host molecules grafted on could affect the mechanical and self-healing property of the hydrogels [81, 82]. β-CD-Ad gels based on different main chains (Poly(acrylamide) (pAAm), poly(N, N-dimethylacrylamide) (pDMAAm), poly(N-isopropylacrylamide) (pNIPAAm), poly (hydroxymethylacrylamide) (pHMAAm), and poly(hydroxyethylacrylate) (pHEA)) were formed. Cycle tensile test revealed different rupture strain and toughness of hydrogels due to different molecular interaction (i.e., hydrophobic interaction, hydrogen bond, etc.) on main chains [81]. A supramolecular hydrogel built on low molecular weight poly(AOI-β-cyclodextrin) (pAOI-β-CD) and polymerizable adamantane monomers (A-TG-Ada) demonstrated brilliant mechanical property. For instance, stretch (>100 times of initial lengths), notch (>50 times of initial lengths) and stab (>30 times of initial lengths) failed to disrupt the integrity of the hydrogel. Self-healing experiment showed similar tensile strain-stress trends between the original and healed hydrogels after 24 h, indicating almost full restoration [82].
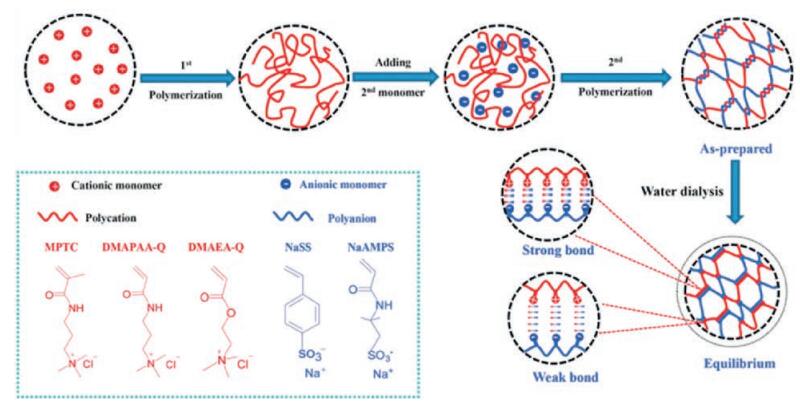
3.1.4. Ionic bondsIonic bonds are formed via the electrostatic attraction between oppositely charged ions. Mixing solutions of oppositely charged monomers or polymers led to formation a hydrogel. The selfhealing mechanism is largely dependent on the migration of the free ions and mobility of the uncross-linked portions of polymer chains, which enable the dynamics and reversibility of the ionic bonding in the 3D network [28]. One of the strategies to the preparation of hydrogels via ionic bonding is the introduction of ferric ions to the system [83-87]. Crosslinking ferric ions with poly (acrylic acid) (PAA) lead to the formation of Fe3+/PAA hydrogel electrolyte [83-85]. The dynamic ionic bonding between carboxylic acid ions on PAA chains and free Fe3+ equipped the hydrogels with self-healing property. When two freshly cut gels were put into contact, the free Fe3+ ions diffused across the interfaces and interact with the carboxylic groups of mobile PAA chains. Selfhealing experiment showed that hydrogels healing for 24 h at room temperature could withstand stretch up to 200% of its initial length and cyclic stretching-relaxation tests revealed the excellent stability of the healed hydrogels even after 1000 cycles [83]. The network in Fe3+/PAA hydrogel could be separated as two parts: the covalently cross-linked PAA chains as the first network and the ionically cross-linked Fe3+/carboxylic acid groups as the second network. Thus, the hydrogels were both mechanically strong and self-healable [84]. Grafting acrylic acid monomers on the surface of vinyl hybrid silica nanoparticles (VSNPs) and subsequent copolymerization and crosslinking yielded Fe3+/VSPN-PAA ionic nanocomposite hydrogels. Improved mechanical strength (tensile strength: 860 kPa, elongation at break: ~2300%) and autonomous self-healing (tensile strength: ~560 kPa, elongation at break: ~1800%) were demonstrated [85]. Similarly, catechol and ferric ions could from ionic bonding [86, 84]. Coordinative crosslinking of catechol-grafted-chitosan and ferric ions lead to hydrogels formation. Electrostatic interactions among the polymer chains provided the hydrogels with load-bearing and self-healing ability. The hydrogels completely regained its original strength within 100 s under cyclic sweep tests (both linear and non-linear time sweeps) [86]. Dopamine, which contains catechol groups, was grafted on montmorillonite (clay) via in situ polymerization [87]. The formed polydopamine-coated clay (D-clay) functioned as the main building block and assembled with ferric ions into three-dimensional hydrogels. The oscillatory rheology test showed that the hydrogels recovered to 70% of its original storage moduli at γ = 0.1% after being placed under large-amplitude oscillatory force (γ = 100%). The ability of self-healing was attributed to the dynamic catechol-ferric ion bonding in Fe3+/D-clay hydrogels. Furthermore, the hydrogels were used as adsorbents to remove Rhodamine 6G (Rh6G) via π-π stacking between aromatic units of polydopamine and Rh6G [87]. Besides ferric ions, carboxybetaine acrylamide (AAZ), a super hydrophilic zwitterionic material, was utilized to construct self-healing hydrogels. The AAZ hydrogels retained 90% of its original tensile strength and modulus after 24 h of healing. Spontaneous self-healing behavior independent of time was also observed in AAZ hydrogels [88]. Recently, Jian Ping Gong and coworkers introduced a general approach to prepare dynamic ionic bonding hydrogels based on the concept of strong/weak bonds. The strong bonds functioned as permanent crosslinking, imparting elasticity, while the weak bonds served as dynamic and reversible sacrificial bonds, imparting self-healing and toughness [18, 28, 29, 89-91] (Fig. 6). Random radical copolymerization of charged-balanced polyampholytes (PAs) carrying opposite charges, followed by dialysis of co-ions and counter-ions, formed PA hydrogels with high toughness (fracture energy ~4000 J/m2) and self-healing ability [89, 90]. Hydrogels healing for 24 h could be stretched up to ~3000% strain, which is close to failure strain of the original hydrogels. The self-healing behavior was greatly influenced by healing temperature, cross-linker density and monomer chemical structure. Increasing the healing temperature (37 ℃ and 50 ℃) and the fraction of weak bonds to total bonds subsequently accelerates self-healing kinetics, whereas more chemical crosslinking and hydrophobic monomers caused a lower healing efficiency [28]. However, the intra-chain ionic bonds gave rise to a globule conformation of the polyelectrolyte chains, which resulted in relatively poor inter-chain entanglement. Consequently, a high preparation concentration was required to form interchain ionic bonds which impart elasticity of the hydrogels [91]. Sequential homopolymerization of oppositely charged polyelectrolytes, however, formed polyion complexes (PIC) hydrogels even at relatively low polymer concentration [29]. The polymers were in extended coil conformation and only inter-chain existed in the network. Thus, PIC hydrogels exhibited more porous and stable structure compared with PA hydrogels. The PIC hydrogels showed enhanced mechanical properties with tensile fracture stress up to 3.7 MPa, modulus of 5.4 MPa, and work of extension reaching 14.8 MJ/m3. Sixty-six percent healing efficiency was reached after 12 h healing in 3 mol/L NaCl aqueous solutions [29].

|
Download:
|
| Fig. 6. Ionic bonding-based self-healing hydrogels. Illustration of the polyion-complex (PIC) hydrogels prepared by sequential homopolymerization of oppositely charged polyelectrolytes. First, the cationic monomer was homopolymerized. Second, the anionic monomer was added into polycation. Third, the anionic monomer was polymerized to form (soft) PIC hydrogels. Fourth, dialysis of the as-prepared hydrogels was carried out to move small counterions and co-ions in the network. As a result, tough PIC hydrogels with equilibrium of weak and strong ionic bonds were yielded. Reproduced with permission [29]. Copyright 2015, WILEY-VCH. | |
{kind=link}
3.2. Dynamic covalent bonds 3.2.1. Imine bonds
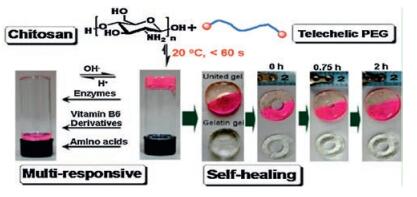
Imine bonds, also known as Schiff base, are the dynamic interactions between amine groups and aldehyde groups [92]. A facial way to prepare imine bonding-based self-healing hydrogels was reported by simply mixing telechelic difunctional poly (ethylene glycol) (DF-PEG) and glycol chitosan in solutions [30, 92] (Fig. 7). Benzaldehyde groups on DF-PEG chains and NH2 groups on chitosan interacted to form dynamic Schiff base linkage. The dynamic equilibrium between the NH2 groups and CHO groups enabled the self-healing ability of the hydrogels. The self-healing tests showed that the punched hole on chitosan/DF-PEG hydrogels disappeared within 15 min and hydrogels being squeezed out of the needle merged into a homogeneous one in 30 min [92]. The chitosan/DF-PEG hydrogels were also responsive to biochemical stimuli such as pH (Schiff base equilibrium), vitamin B6 derivative (chitosan competitor), amino acids (aldehyde competitor) and enzymes like pepsin [30]. Biocompatibility was demonstrated by encapsulating HeLa cells in 3D network and the cells remained ~87% viability after 72 h culturing [92]. By introducing carboxy modified Fe3O4 nanoparticles into chitosan/DF-PEG hydrogels, a magnetic-responsive phenomenon was observed. The broken pieces of hydrogel gathered together to heal and the hydrogels could squeeze through a narrow channel integrally under magnetic field [93]. However, the chitosan/DF-PEG hydrogels possessed a weak mechanical strength. For example, the storage moduli G′ was only around 1 kPa, which limited its biomedical application. Lina Zhang and coworkers improved G′ to ~3160 Pa by adopting Benzaldehyde-terminated telechelic four-armed polyethylene glycol (PEG-BA) and carboxymethyl chitosan (CMC) as the reaction reagents [94]. Unlike conventional two-arm linear PEG, the fourarm PEG behaved like impenetrable space-filled spheres, which formed more homogeneous network with reduced polymer entanglements and loops. Therefore the four-armed PEG held a greater potential to form tough hydrogels. The CMC/PEG-BA hydrogels were also used in vivo as hemostatic materials in rabbit bleeding liver models [94].

|
Download:
|
| Fig. 7. Imine bond-based self-healing hydrogels. Schematics and photographs chitosan/DF-PEG hydrogels. The hydrogel formed via interaction between benzaldehyde groups on DF-PEG chains and NH2 groups on chitosan demonstrated self-healing ability (5% gelatin gel as control) and multi-responsiveness to external stimuli (pH, vitamin B6, amino acids and enzymes). Reproduced with permission [30]. Copyright 2011, American Chemical Society. | |
{kind=link}
Apart from functionalized PEG, various polymers with aldehyde groups could interact with chitosan to form a dynamic imine bonding-based self-healing hydrogels. Different composite material contributed to unique property and application of the resultant hydrogels. For instance, chondroitin sulfate multiple aldehyde (CSMA) interacted with N-succinyl-chitosan (SC) to form an injectable self-healing hydrogel [31]. Both CSMA and SC were natural polymers with good biocompatibility, biodegradability and nontoxicity. Subcutaneous injection of the hydrogels into rats aroused little inflammation and enzymes like PH-20, hyaluronidase and lysozyme degraded the hydrogels in several weeks in vivo [31]. Hydrogels prepared by oxidized alginate and acrylamidemodified chitin (AMC) were able to guide the reconstruction of inorganic biomaterial like hydroxyapatite (HA). The separated pieces of hydrogels with HA could heal autonomously when put together. Furthermore, the lyophilized HA hydrogels retained selfhealing property when being rehydrated in water [95]. Employing hydrogels to adsorb nuclear waste might seem unique, yet it was realized by Schiff-base-based self-healing hydrogels [96]. Chitosan/formaldehyde based hydrogels were utilized to isolate fission products 152Eu (T1/2 = 13.33 a) and 137Cs (T1/2 = 30.17 a) in aqueous HCl medium using solid liquid extraction technique. The dynamic chitosan-based hydrogels was featured as the solid phase, providing high surface area to actively interact with and porous structure to accommodate 152Eu [96]. Tetra-aldehyde functionalized zinc phthalocyanines (TA-ZnPc), a light-absorbing molecule, cross-linked with chitosan to form a hydrogel which showed great promise in photodynamic therapy (PDT) of cancer [97]. Zinc phthalocyanines were responsive to light irradiation (wavelength ~670 nm) and generated singlet oxygen (1O2) which killed tumor cells. Conjugation of TA-ZnPcs to hydrogels via Schiff-base reaction gave PDT unexpected outcome. The hydrogels were injectable and self-healable, so in situ injection around tumors and simultaneous self-healing avoided adverse effect on normal tissues. Besides, the hydrogels were pH sensitive and biodegradable, which meant the controlled-release of TA-ZnPc was triggered only in acidic tumor environment [97]. Incorporation of multiwalled carbon nanotubes (MWCNTs) into chitosan/TA-ZnPc system imparted the hydrogels with improved mechanical (G′ = ~22.4 kPa, 12 wt% TA-ZnPc, 2 wt% MWSNTs) and electrical properties. The hydrogel with MWSNTs (2 wt%) were able to conduct adequate physiological electrical stimulus (2.94 10×-3 S/cm) on cells, indicating its potential biomedical application [98].
Chen et al. developed a hydrogel based on aldehyde modified xanthan gum (ALD-XA) and phosphatidylethanolamine (PE) liposomes Schiff base linkages [99]. The hydrogels were responsive to temperature, pH variation and biological stimuli (histidine). The self-healing tests revealed that two cut pieces of hydrogels could merge together within 15 min under ambient conditions. The hydrogels were injectable and biocompatible, so it could be used as cell carrier for 3D culture [99].
3.2.2. Acylhydrazone bondsAcylhydrazone bonds are dynamic covalent bonds based on reaction of aldehyde and hydrazine groups, which were feasible to prepare self-healing hydrogels [100, 101]. This strategy was developed by condensation reaction between acylhydrazines at the two ends of a poly(ethylene oxide) (PEO) and aldehyde groups in tris[(4-formylphenoxy)methyl] ethane catalyzed by acetic acid [19, 102] (Fig. 8a). The hydrogels exhibited reversible sol-gel transition properties by changing pH of the system. Two cut hydrogels merged together and could be lifted by tweezers when kept in contact without any other intervention for 7 h [102]. In order to clarify the molecular mechanism underlying mechanical and self-healing properties, a serial of features like gelation kinetics and rheological behaviors were examined [19]. Similarly, a copolymer of N-isopropylacrylamide and acylhydrazine P(NIPAMco-AH) (PNIPAM) crosslinked with PEO dialdehyde to form selfhealable hydrogels [32]. Excessive acylhydrazine groups in hydrogels activated and facilitated the self-healing procedure. The hydrogels healing for 24 h could not be split along the cutting line when stretched. Furthermore, the hydrogels showed thermoresponsiveness due to temperature sensitivity of PNIPAM content [32]. Hydrogels formed by reaction of hyperbranched poly(amido amine) (HPAMAM) and oxidized alginate (OALG) also displayed a fluorescent property besides self-healing ability and pH responsiveness. The hydrogels emitted blue light under UV irradiation (365 nm) [103].

|
Download:
|
| Fig. 8. Acylhydrazone bond-based self-healing hydrogels. (a) Schematic illustration of condensation reaction between acylhydrazines at the two ends of poly(ethylene oxide) (PEO) and aldehyde groups in tris[(4-formylphenoxy)methyl] ethane at a slightly acidic environment (pH 4.0-6.0). When pH < 4, the cross-linked network disassembled. Reproduced with permission [102]. Copyright 2010, American Chemical Society. (b) Schematics of chemical structure of HG1G2 hydrogels. Both acylhydrazone bonds (under acidic conditions) and disulfide bonds (under basic conditions) contributed to the self-healing of HG1G2 hydrogels. Reproduced with permission [37]. Copyright 2012, American Chemical Society. | |
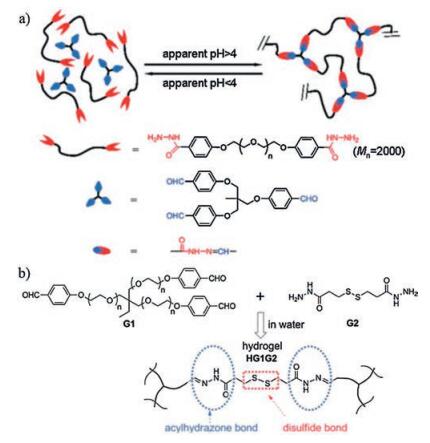{kind=link}
However, it should be noted that the reversibility of acylhydrazone bonds is limited to a slightly acidic environment (pH 4.0-6.0), which means thus formed hydrogels can only proceed the healing procedure within a narrow pH range [32, 102]. To solve this defect, Yongming Chen and coworkers incorporated dual-dynamic bonds into one system. By simply mixing aldehyde-terminated tris-armed PEO (G1) and dithiodipropionic acid dihydrazide (G2), a self-healing hydrogel (HG1G2) containing acylhydrazone and disulfide bonds were formed [37] (Fig. 8b). HG1G2 hydrogels could selectively chose specific healing routes according to different pH environments. In acidic conditions, the reversible acylhydrazone bonding mediated the self-healing, yet in alkaline conditions, disulfide exchange reaction was active. Self-healing experiments showed that the cut segments integrated into one plate automatically when kept in contact for 48 h with pH either being 6 or 9. However, in neutral conditions (pH 7), self-healing failed because both acylhydrazone and disulfide bonds are "locked". Addition of aniline functioning as acylhydrazone exchange reaction catalyst helped with the healing under neutral condition, with >50% healing efficiency after 48 h [37]. Based on the same idea of incorporating dual-dynamic bonds into one system, a biocompatible CEC-I-OSA-I-ADH hydrogel was designed, in which N-carboxyethyl-chitosan (CEC) and adipic acid dihydrazide (ADH) reacted with oxidized sodium alginate (OSA) respectively by dynamic imine and acylhydrazone bonding [33]. Under neutral conditions, imine exchange reaction played the dominance, imparting self-healing. Up to 90% ± 2.7% healing efficiency was achieved after healing for 48 h at 25 ℃ as demonstrated by beam-shaped compression test. Alternate step strain rheological measurement (γ = 1%, 80%, 300%, and 800%) and macroscopic injection-and-healing experiment further characterized the immediate and efficient healing. The hydrogels also displayed great cytocompatibility as the encapsulated cells maintained 95.3% ± 3.4% viability after 48 h culture as demonstrated by Live/ Dead staining [33].
3.2.3. Boronate ester bondsThe dynamic covalent boronate ester bonds exist in complexations of certain diols and boronic acid [104]. Inspired by mussel adhesive proteins, catechol is one of the diols frequently utilized in the fabrication of boronate ester bonding-based self-healing hydrogels apart from its application in H-bonding-based hydrogels (described in the part of "hydrogen bonds") [35,]. Messersmith and colleagues prepared hydrogels via interaction between catechol derived four-arm PEG (cPEG) and 1, 3-benzenediboronic acid (BDBA) at 20 ℃ [35]. The hydrogel was pH-sensitive, which could only be produced and stabilized in alkaline condition. When the hydrogels were exposed to acidic environment (pH 3), the network was disturbed and sol state was generated (Fig. 9a). The favorable pH values required for the stabilized hydrogel formation were around 9, which is lower than the pKa of catechol (pKa = 9.3) and higher than the pKa of boronic acid groups (pKa = 8.7). When two cut pieces of hydrogels were put in contact, they fused together within 30 s and could resist stretching, which is due to the dynamical reaction between free boronic acid and catechol (charaterized by 11B NMR). The hydrogels almost recovered to initial G′ in 100 s at 5% strain after being placed under 1000% strain as rheological measurements demonstrated [35]. Another mussel-inspired polymer, poly (dopamine methacrylateco-N-isopropylacrylamide) (p(DMA-co-NIPAM)), was able to interact with boric acid (H3BO3) to form self-healing hydrogels. In addition to pH sensitivity, the hydrogel also exhibited thermo-responsiveness due to the DMA and NIPAM monomers. The lower critical solution temperature (LCST) of the polymer solution was pH-and DOPA-content-dependent [105].

|
Download:
|
| Fig. 9. Boronate ester bond-based self-healing hydrogels. (a) Schematic illustration of pH-responsiveness of the hydrogel formed via dynamic interactions between cPEG and 1, 3-benzenediboronic acid (BDBA). Reproduced with permission [35]. Copyright 2011, The Royal Society of Chemistry. (b) Chemical structure of intramolecular coordination B-O bond formed in monomer 2APBA, which can strengthen the boronic acid-diol complexation. Reproduced with permission [36]. Copyright 2015, American Chemical Society. | |
{kind=link}
The properties (e.g., pH-sensitivity, thermal-responsiveness) of boronate ester bonding-based hydrogels were tunable by changing internal structures and external environment [36, 41]. For instance, by introducing 2-acrylamidophenylboronic acid (2APBA) as the reaction monomer, the obtained hydrogels remained stable even at neutral and acidic pH [36]. This is because an intramolecular coordination between carbonyl oxygen of its acrylamido moiety and boron of the boronic acid group was formed in monomer 2APBA, which is believed to strengthen the boronic acid-diol complexation (Fig. 9b). Oxidation of the catechol groups in hydrogels reduced the self-healing ability and creep but led to increased stiffness. Besides oxidation, external environment change like temperature also affected hydrogels' properties. Gosecka et al. studied the effect of thermal effects on hydrogels [41]. They prepared hydrogels using hyperbranched polyglycidol (HbPGL) and borax as monomers, then the molecular structure and cross-linking density were measured using tools like solid-state boron nuclear magnetic resonance (11B NMR), positron annihilation lifetime spectroscopies (PALS) and rheological test. Heating (-10 ℃ to +70 ℃) not only reduced the average cross-link lifetime of the hydrogels, but also decreased the fraction of boronic diester in hydrogel system and released HbPGL macromolecules from the network [41].
Recently, strategies were developed to improve the applicability of boronate ester bonding-based self-healing hydrogels in different fields [106-110]. One is the use of glucose. Glucose is another type of diols which could form a stable five-membered ring complex with phenylboronic acids [106, 107]. Hydrogels adopting boronic acid-glucose ester complexes displayed shearthinning and self-healing properties [106]. Based on the idea that glucose could competitively bind to boronic acid in other boronic acid-diols systems, it is plausible to utilize free glucose to control the drug release behavior of hydrogels. Anderson's group designed a PEG-based hydrogel network (PEG grafted with phenylboronic acid derivatives and cis-diols) where insulin and globulin G (IgG) were respectively incorporated [107]. Free glucose in solution disrupted the hydrogel network, thereby changing the mesh size of the hydrogels and accelerating the release kinetics of IgG (13% released in glucose-free buffer, 46% released in 4 mg/mL glucose buffer over 48 h). However, release kinetics of insulin was hardly affected, for its poor sensitivity to mesh size of the hydrogels [107]. Besides the use of glucose, nanomaterial graphene oxide was incorporated into the hydrogels to enhance mechanical properties (10-fold increase in storage moduli of hydrogels with GO) [108]. Hydrogels from phenylboronic acid grafted alginate (Alg-PBA) and PVA exhibited shape-memory capability. Apart from its reaction with PVA, Alg-PBA also formed network with Ca2+, which could be extracted from the hydrogels and thus contributed to the shape memory ability [109]. A facile and economical one-pot approach was developed to fabricate PEG-based self-healing hydrogels from thiol-end click and boronic acid-diols chemistry [110]. Poly (ethylene glycol) diacrylate (PEGDA), dithiothreitol (DTT), and borax were added into one pot, where borax both formed borax diol complexation (with borax and DTT) and catalyzed Michael addition (between PEGDA and DTT). The hydrogels exhibited rapid gelation (from 40 s to 2 min), good mechanical strength (G′ up to 104 Pa), self-healing (within 30 min), pH-and thermal-responsiveness [110].
3.2.4. Other dynamic covalent bondsDiels-Alder reaction is a "click" reaction between a conjugated diene and dienophile [111, 112]. Using Diel-Alder reaction to fabricate self-healing hydrogels is promising for its advantages like high reaction selectivity, no side reactions, reversibility, etc. [13, 113-115]. A cytocompatible dextran-based self-healing hydrogel was prepared via Diels-Alder reaction between monomers fulvene-modified dextran and dichloromaleic-acid-modified poly (ethylene glycol) at physiological conditions [13] (Fig. 10a). Selfhealing ability was measured visually and by rheological tests. Continuous step strain test revealed that G′ of the hydrogels could repeatedly retain its initial value at 5% strain within 300 s after being treated with 1000% strain. Two cut hydrogels merged into an undistinguishable one after being put into contact for 12 h at 37 ℃ without any other intervention, and withstood its own weight when lifted. Scanning electrochemical microscopy (SECM) was utilized to monitor in situ topography of the healing interfaces. It was shown that both longitudinal depth of the scratch disappeared after 7 h, and the healing ratio was calculated to be 98.7% as compared the scratch depth with the original unhealed one [13].

|
Download:
|
| Fig. 10. Self-healing hydrogels formed via reversible Diels-Alder reaction and disulfide bonding. (a) Chemical structures of the dextran-based self-healing hydrogel formed via fulvene groups on dextran and dichloromaleic acid groups on PEG at physiological conditions. Reproduced with permission [13]. Copyright 2013, WILEY-VCH. (b) Schematic illustration of crosslinking structure of Au-S/S-S based self-healing hydrogels. Au(I) capping strategy protected thiol from aerial oxidation. Reproduced with permission [118]. Copyright 2015, American Chemical Society. | |
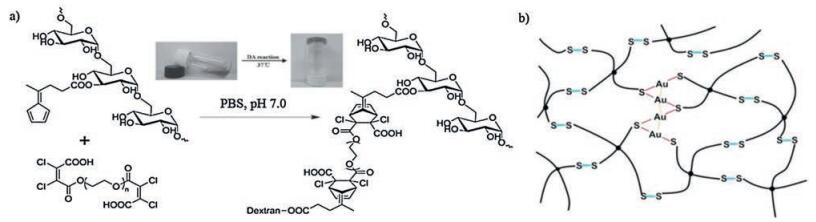{kind=link}
Disulfide bonds are based on thiol/disulfide exchange reaction which occurs in the presence of nucleophilic thiolates at neutral or alkaline environment [116]. The mild reaction condition (ambient environment at low temperature) imparts its potential use as conjugating mechanism in self-healing hydrogels [117]. A selfhealing hydrogel coating, based on thiol/disulfide exchange reaction, was developed via thiol-ene photopolymerization of poly(ethylene glycol)methyl ether methacrylate (PEGMA), N-hydroxyethyl acrylamide (HEAA), 2-(methacryloyloxy)-ethyl trimethylammonium chloride (META) and bis(2-methacryloyl)-oxyethyl disulfide (BMOD) on stainless steel surface [14]. BMOD contains disulfide bonds which facilitate self-healing. Optical microscope and atomic force microscope (AFM) were harnessed to monitor self-healing process. The results showed that when a "mobile phase" was created with a bit of water dropped at the interfaces, the size of the cut scratch recovered from 7.8 mm to 4.2 μm in width after 12 h. Due to the functional monomers utilized, the hydrogels possessed antifouling and antibacterial properties (reduced bovine serum albumin and E. coli adhesion) [14]. Nevertheless, it should be noticed thiol would be oxidized into disulfide when exposed to air. Thus, the disulfide bondingbased self-healing hydrogels were assumed to lose dynamic properties (e.g., self-healing behavior) in the long run. To address this issue, Damien Dupin and coworkers put forward a metal(Ⅰ) [Au (Ⅰ) or Ag(Ⅰ)] capping strategy to protect thiol from aerial oxidation [118] (Fig. 10b). They fabricated hydrogels by simply mixing tetraarm thiol-terminated PEG homopolymer [(PEGSH)4] with HAuCl4/ AgNO3 solutions at room temperature. Au/Ag(I) capping shields thiol from oxidation and the presence of Au/Ag-S and S-S exchange reaction imparts the dynamic properties of the hydrogels. Rheological test showed that two cut pieces of hydrogels healing for 1 min achieved almost complete recovery. Besides, the shape of the hydrogels could be changed within hours when put into different molds. The hydrogels also displayed low cytotoxicity as demonstrated by cell encapsulation [118]. Afterwards, the same group incorporated agglomerated bioactive glass nanoparticles (10 μm clusters) (BAG) into the network to form Au-(PEGSH)4-BAG hydrogels [119]. The addition of BAG nanoparticle not only improved the mechanical properties of the hydrogels (though the dynamic character was slightly diminished), but also exhibited osteo-inducing effects via the formation of hydroxyapatite as a result of BAG degradation. Furthermore, the hydrogels retarded the degradation of the BAG nanoparticles, thereby maintaining the physiological condition of the aqueous solution and increasing cytocompatibility of BAG nanoparticles [119].
Oxime bonds, belonging to a type of imine bonds, are formed by dynamic covalent reaction between hydroxylamine and aldehydes/ketones groups [120]. Oxime bonding reaction is considered to be an ideal conjugating way to synthesize hydrogels because it possesses advantages like high reaction efficiency, mild reaction conditions, harmless side products, and generally greater stability compared with imine bonds [120]. Recently, an oxime bonding-based self-healing hydrogels were fabricated via radical polymerization of N, N-dimethylacrylamide (DMA) and diacetone acrylamide (DAA), which then cross-linked with difunctional O, O′-1, 3-propanediylbishydroxylamine dihydrochloride [38] (Fig. 11). Oxime exchange reaction was triggered by excessive alkoxyamines which contributed to the dynamic nature of the hydrogels, namely self-healing properties. Step-strain measurements showed that hydrogel transited from sol to gel state in less than 17 s (G′ > G″) at 50% strain after it was treated with 1000% strain. Visually, two cut pieces of hydrogels healing for 2 h resisted stretching and withstood its own weight, and this process was repeatable [38].

|
Download:
|
| Fig. 11. Self-healing hydrogels formed via dynamic oxime bonds. Schematics and photographs showed the chemical structure of P(DMA-stat-DAA), which could crosslink with difunctional alkoxyamines O, O′-1, 3-propanediylbishydroxylamine dihydrochloride to form hydrogels. Reproduced with permission [38]. Copyright 2015, The Royal Society of Chemistry. | |
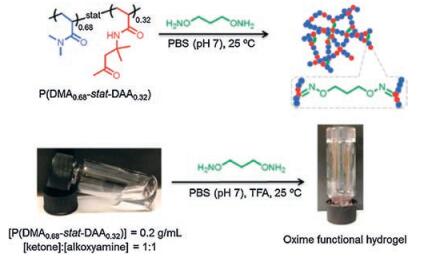{kind=link}
4. Biomedical application of self-healing hydrogels
Hydrogels are promising biomaterials for cell therapy, tissue engineering and drug delivery [1, 121]. However, most hydrogels are hydrophilic cross-linked three-dimensional system susceptible to external stimulus. Once the structural integrity is disrupted by either the external mechanical force or the physiological erosion, the spatiotemporal functioning mode of hydrogels is lost. Thus, numerous strategies have been developed to facilitate hydrogels with better bio-mimic ability, one of which is the design of selfhealing ability in hydrogels system [9]. With the ability to self-heal, hydrogels can repair themselves in response to damage and recover their original structures and properties, consequently improving the reliability and safety to accomplish their functions in predetermined way [9, 122, 123]. Although self-healing is one of the fundamental properties of living tissues and strategies to design self-healing hydrogels have been extended developed, the biomedical application of self-healing hydrogels are rarely reported. In this section, we mainly focus on the progress being made in the biomedical application of self-healing hydrogels.
To address biomedical usage, the premise is hydrogels are not supposed to exhibit toxicity to cells and tissues. Before in vivo embedding, in vitro cytotoxicity tests are conducted. Encapsulation of cells into hydrogels is a direct way to test the biocompatibility of the self-healing hydrogels. Live/Dead staining helps to visualize the viability and morphology of cells in the three-dimensional culture system under confocal microscope [92, 107]. Alternatively, hydrogel leachate can be used as cell culture medium and the cell viability and proliferation are tested to determine the biocompatibility of self-healing hydrogels [124]. Usually, hydrogels fabricated from natural polymers were more biocompatible than those from synthetic polymers.
For the in vivo application, the constituents (monomers), structures (e.g., porosity, mesh size, network alignment), and the mechanical properties of self-healing hydrogels are essential, which might not only improve the self-healing ability, mechanical properties of self-healing hydrogels, but also facilitate the biocompatibility and optimize the tissue-specific in vivo application [15, 125-128]. Nowadays, self-healing hydrogels were mainly harnessed in tissue engineering and drug delivery related fields.
In the field of cartilage and bone tissue engineering, self-healing hydrogels had an excellent performance [15, 125, 128]. For instance, a cartilage tissue-adhesive self-healing hydrogel was developed based on Diels-Alder reaction and acylhydrazone bonds [15]. As is known, hydrogels are considered as good cartilage tissueengineered scaffolds. However, when implanted, hydrogels displaying poor integration to the surrounding cartilage tissue showed increased risk in the looseness and fracture of the structure even under gentle daily movement. With the reversible acylhydrazone bond, the intelligent autonomous self-healing property has been introduced, and good tissue-adhesive property with the local cartilage tissue has been achieved. The adhesion resulted from the dynamic Schiff-base reaction between -CHO groups on the hydrogel monomers (aldehyde-functionalizedhyaluronic acid-furylamine (HA-furan-CHO)) and the amide groups on cartilage, and the adhesive strength could reach to 10.3 ± 0.7 kPa [15] (Fig. 12a, b). In bone tissue engineering, the introduction of reversible bonds was considered to assemble smart, functional materials. A hydrogel exhibiting mineral surface adhesiveness and bone interactive capacity were formed via noncovalently crosslinking of calcium phosphate nanoparticles and bisphosphonate-functionalized hyaluronic acid [125] (Fig. 12c). This non-covalent cross-linking would be sufficiently strong to resist the highly dynamic in vivo conditions. In vivo subcutaneous implantation showed that this self-healing hydrogel could retained their integrity and microstructure well, while little cell infiltration in central regions was aroused after 4 weeks, demonstrating good biocompatibility. Moreover, when embedded in rats bone defect, the hydrogels supported bone conduction and promoted the homogenous formation of trabecular-like bone throughout the defects [125] (Fig. 12d). Hydrogel based on the above reversible bonds aids in the progression of bone formation throughout the material and is particularly appealing for bone regenerative applications [125].

|
Download:
|
| Fig. 12. Self-healing hydrogels used in cartilage and bone research. (a) Schematic of the interfacial interaction between HA-furan-CHO motifs in hydrogels and cartilage surface via Schiff-base reaction. (b) SEM observation of the interfaces between hydrogel and cartilage. Diels-Alder based non-adhesive hydrogels showed no integration with cartilage (b1, b2). Diels-Alder and acylhydrazone bonds-based adhesive hydrogel exhibited apparent integration with cartilage (b3, b4). Reproduced with permission [15]. Copyright 2015, American Chemical Society. (c) Schematic of self-healing hydrogels via non-covalently crosslinking of calcium phosphate (CaP) nanoparticles and bisphosphonate-functionalized hyaluronic acid. (d) the HABP-CaP hydrogels supported bone conduction and promoted the homogenous formation of trabecular-like bone throughout the defects after 4 weeks of implantation in bone tissue. Reproduced with permission [125]. Copyright 2014, Elsevier Ltd. | |
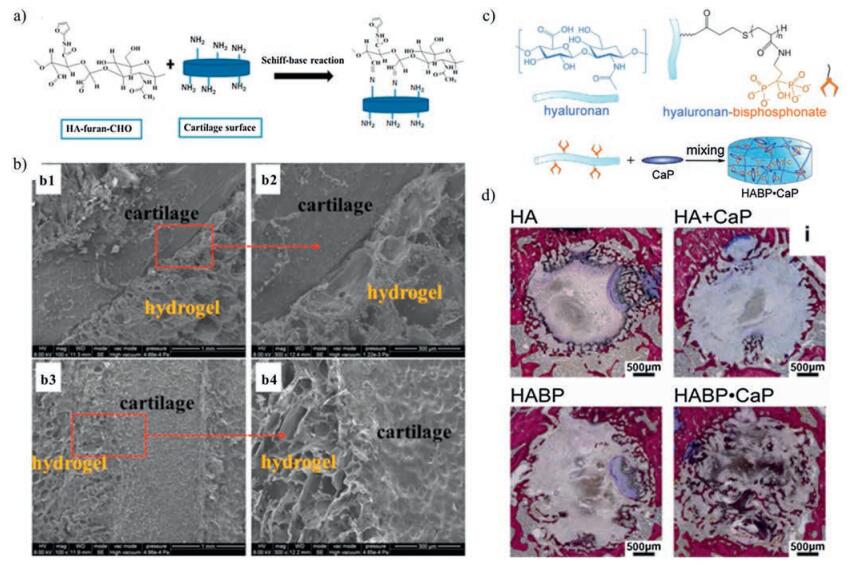{kind=link}
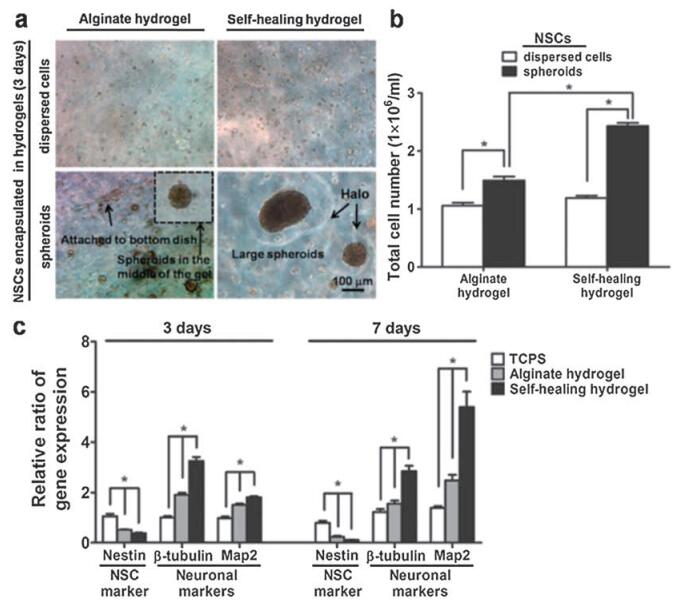
In the field of neural tissue engineering, self-healing hydrogels with appropriate mechanical properties are able to mimic the biomechanical signals of extracellular matrix (ECM), functioning as ideal 3D culturing matrix for cell proliferation/differentiation and scaffolds for tissue engineering [126]. A chitosan-based selfhealing hydrogel with proper modulus (~1.5 kPa stiffness) was shown to assist in healing of central nervous system deficits (Fig. 13). Moreover, in zebrafish embryo neural injury model, the self-healing hydrogels were shown to rescue the neural injury (38% recovery with hydrogels alone, 81% recovery with neural stem cell spheroids encapsulated in hydrogels). Moreover, the self-healing properties made the hydrogel more injectable. Non-healable hydrogels suffered from the compressive strain during injection and resulted in fracture and structural damage, while the selfhealing hydrogel can quickly resume the original state upon strain removal [126].

|
Download:
|
| Fig. 13. Chitosan/DF-PEG self-healing hydrogel with proper stiffness assisted in healing of CNS deficits. (a) Photographs of dispersed NSCs and NSC spheroids encapsulated in the alginate or the self-healing hydrogels for 3 d. "Halo" was observed for NSC spheroids in self-healing hydrogels. (b) Total cell number of NSCs and NSC spheroids in alginate or self-healing hydrogels after 3 d. (c) Real-time RT-PCR showed the relative ration of specific neuronal-related genes expression after 3 d and 7 d. The expression was normalized to TCPS (3 d). *p < 0.05. Reproduced with permission [126]. Copyright 2015, WILEY-VCH. | |
{kind=link}
In the field of cardiovascular tissue engineering, and the implantable or wearable biosensors for human health monitoring, conductive hydrogels are of promise. Meanwhile, with the ability to response to damage and recover, self-healable hydrogels can consequently improve the reliability and safety during their functional lifetimes. It is a tough job to construct the conductive hydrogel with both outstanding mechanical property and selfhealable property. Studies showed that polymerized acrylamide (PAM) interacted with polymerized dopamine (PDA) and partially reduced graphene oxide (pGO) via mussel-inspired non-covalent interactions to form PDA-pGO-PAM self-healing hydrogels [127]. The well-dispersed reduced GO (rGO) in the network intertwined to form electronic pathway, imparting conductivity. Thus the hydrogels served as ideal in vivo electrodes/stimulators and detectors. The dorsal muscle implantation in rabbits exhibited the greater sensitivity of hydrogels to electrical signals in deep tissue than commercially available surface electrodes [127]. Besides, good cell adhesiveness of this self-healable hydrogel was demonstrated because of non-covalent interactions between catechol groups on PDA and amine/thiol groups on cell membrane. Another conductive self-healing hydrogel was developed by dynamic imine bonding (Schiff-base reaction) between chitosangraft-aniline tetramer (CS-AT) and dibenzaldehyde-terminated poly(ethylene glycol) (PEG-DA) [128]. The conductivity of the hydrogels ranged from 2.29 10×-3 S/cm to 2.42 10×-3 S/cm, similar to that of cardiac tissue (1-4 10×-3 S/cm), indicating its potential use in cardiac repair. Besides, the hydrogels displayed enhanced antibacterial ability (E. coli and S. aureus) due to the positive charged amino groups on CS-AT chains. Good tissue adhesiveness (from 3.1 ±0.69 kPa to 5.1 ±0.83 kPa), injectability, and biodegradability were demonstrated too [128].
The self-healing performance of hydrogels is helpful to tissue repair, especially with drug delivery capacity. Drug delivery and its control methods are the area of intense research and the central tenet of tissue engineering. An approach to this area is to use scaffold biomaterials to deliver and release therapeutic drugs in a sustained manner. Pharmacologic therapies for disease are limited by traditional delivery routes. Self-healing hydrogel with its advantageous ability of resisting strain and recovering could maintain and benefit the drug delivery system. Drug loading and releasing ability of self-healing hydrogels is what has been exploited in vivo [17, 129]. Meanwhile, whether the self-healing ability of hydrogels would be affected by the addition of drugs was also explored. For example, colistin, a lipopeptide antibiotic, was incorporated into glycol chitosan/DF-PEG self-healing hydrogels via dynamic imine bonding [129]. The amount of colistin loaded was tunable, while the structure and mechanical properties of the self-healing hydrogels were not compromised by colistin. Moreover, in the in vivo application to a mouse burn infection wound, the hydrogels achieved localized stable release of colistin, resulting in up to 99% bacteria killing even including colistin resistant P. aeruginosa strains [129]. Self-healing hydrogels employing polymer-nanoparticle interactions succeed in dual loading of hydrophobic and hydrophilic drugs in one system. The polymernanoparticles (PNP) hydrogels were formed by non-covalent interaction between hydroxypropylmethylcellulose derivatives (HPMC-x) and poly (ethylene glycol)-block-poly(lactic acid) (PEG-b-PLA) NPs [17] (Fig. 14). Different drugs could be encapsulated in the PNP hydrogels simultaneously yet the release profile was independent. The hydrophilic drugs exhibited first-order release (1-4 days), while the hydrophobic drugs showed burst release followed by zero-order release (>30 days). In vivo test further proved the biocompatibility of the hydrogels, and a simultaneous release of hydrophilic and hydrophobic drugs model was successfully built [17]. Glucose-controlled release of incorporated molecules (IgG) were developed in PEG-based hydrogel network. Free glucose in solution disrupted the hydrogel network, thereby changing the mesh size of the hydrogels and accelerating the release kinetics of IgG (13% released in glucose-free buffer, 46% released in 4 mg/mL glucose buffer over 48 h) [107]. Hydrogels with controlled proteolytic degradation were attained by Jason et al. [72]. Another hydrogel was formed by the host-guest interactions between β-CD and Ad. A peptide tether interlinked βCD and Ad to the backbone (hyaluronic acid). When injected subcutaneously, the peptide could be hydrolyzed by protease (MMP-2 and collagenase), and the hydrogels were subsequently degraded. Moreover, by controlling the amino acid residues of peptides, the degradation kinetics of the hydrogels could be further tailored and used in the controlled drug delivery system [72]. Adopting peptides as the building block to form self-healing hydrogels were achieved [130]. The 9-residue amphiphilic peptides (N-Pro-Ser-Phe-Cys-Phe-Lys-Phe-Glu-Pro-C) self-assembled as nano-fibers, which further overlapped to form self-healing hydrogels at high concentration. The amphiphilic nature of the peptide hydrogels made it possible to load hydrophobic drugs (e.g., pyrene) yet remain aqueous solubility. Beside, in vivo application showed the hydrogels were capable of accelerating the hemostatic process in rat liver bleeding model [130]. Interestingly, hydrogels developed from the therapeutic agent itself as the monomer was reported [131]. Combination of calcium ions and steroid drugs (dexamethasone phosphate (DexP), betamethasone phosphate (BetB), or hydrocortisone phosphate (HydP)) via coordination interactions led to the formation of steroid-drug-based selfhealing hydrogels. The drug release kinetics was tunable by changing molar ratio of drugs and Ca2+. In vivo application revealed that DexP hydrogels alleviated inflammation induced by bupivacaine and prolonged the sensory nerve block effect of sciatic nerve (twice longer than bupivacaine alone). Besides, the hydrogels were degraded within 8 days and showed no visible residue when drug release was finished [131]. An ultrasound-triggered drug delivery strategy to improved chemotherapy was developed in an ironically cross-linked self-healing hydrogel [12]. The hydrogels cross-linked from alginate and Ca2+ could be disrupted by ultrasonic and reversibly reformed at the presence of physiological level of Ca2+. Mitoxantrone was incorporated into the hydrogels and demonstrated ultrasound-triggered "burst" release. In vivo treatment of xenograft human breast tumors in nude mice showed that ultrasound-triggered drug release inhibited tumor growth and induced apoptosis to a greater extent compared with systemic drug delivery in the short term. Though no significant improvement in survival was observed in the long-term study, the tumor size was greatly reduced in ultrasound treated group [12].

|
Download:
|
| Fig. 14. Dual-therapeutic release of polymer-nanoparticle (PNP) self-healing hydrogels. (a) A simultaneous release of hydrophobic (TR-loaded NPs; top) and hydrophilic (BSAAF; bottom) therapeutic model was successfully built in vivo, as demonstrated by intravital fluorescence imaging. (b-c) Plots of relative intensity of fluorescence versus distance from the center (n = 5) at 1 h (b) and 12 h (c). (d) Distance of therapeutic release at 20% relative fluorescence intensity over time (n = 5). Reproduced with permission [17]. Copyright 2015, Macmillan Publishers Limited. | |
{kind=link}
Other biomedical application was also developed to strength the in vivo performance of self-healing hydrogels [94, 132, 133]. Hydrogels prepared from poly (N-isopropylacrylamide) (PNIPAM) coated with polydopamine nanoparticles (PDA-NPs) exhibited near-infrared (NIR)-responsiveness and cell/tissue adhesiveness (adhesion strength to porcine skin up to 90 kPa as demonstrated by tensile-adhesion test). The PDA-NPs hydrogels loaded with EGF displayed wounding healing ability in a rat model [132]. In vivo visualization by magnetic resonance imaging (MRI) was one way to monitor the biodegradation profiles of self-healing hydrogels [133]. A Gd (Ⅲ)-binding hydrogel was developed via dynamic imine binding of Gd (Ⅲ) chelated-diethylene triamine pentaacetic acid (DTPA-Gd) and chitosan. A strong signal on T1-weighted MRI images was detected when Gd3+-hydrogels were injected subcutaneously. Moreover, the hydrogels could be continuously detected by MRI until the biodegradation was complete [133].
5. Conclusion and future prospectsGreat advances have been made in the field of self-healing hydrogels. Yet the extensive application of self-healing hydrogels still faces great challenges. General and practical standards with parameters should be developed to fully characterize and describe the self-healing process and outcome. Standardized testing methods regarding morphological/topographical observation and mechanical characterization of self-healing hydrogels need to be developed. Morphologically, multi-scale characterization is required, covering from atomic-scale, molecular-scale to macroscopic-scale. Atomic-and molecular-scale observation give more details about the healing process (e.g., structural integrity and conformity after healing) and may help to illustrate the healing mechanism apart from theoretic deduction. Also, how to translate these visual and qualitative information into numerical and quantitative datum remains to be answered. Mechanical characterization is now mainly employed to calculate the healing efficiency and healing ratio in a short time. Yet the long-term changes should be considered. Besides, methods to numerically demonstrate healing kinetics need to be further explored.
To develop the optimal self-healing hydrogel system, further researches are need such as incorporating excellent mechanical properties and efficient self-healing ability into one system, achieving good biocompatibility and unique bio-functionality in sophisticated bio-environment. Hydrogels with multi-responsiveness, improved mechanical properties and biomimetic are to be continually studied. Besides, as self-healing hydrogels were mainly cross-linked by polymers, low molecular weight (LMW) self-healing hydrogels with typical thixotropic feature should be further developed. While great advancement has been made for "tough" hydrogels such as interpenetrating polymer network [74] and nanoparticles [17], etc., biomedical application of the selfhealing hydrogels just started. Nowadays, the advantage of selfhealing ability compared with non-healable one in biomedical application has not been verified yet because studies using selfhealing hydrogels in vivo failed to set a non-healable hydrogel group control. Thus, whether hydrogels with self-healing ability are superior and the extent to which self-healing ability contributes to tissue repair, tumor inhibition and sustained drug releasing must be further investigated. To achieve optimal biomedical application, the design of the self-healing hydrogels should be based on the presumptive application. Future design and biomedical application of hydrogels could refer to the following suggestions: (1) biocompatibility and biodegradability of the hydrogels could be improved; (2) hydrogels should heal autonomously without external stimuli. Based on the prerequisite, hydrogels with stimuli-triggered healing or responsiveness could be further tailored to meet additional demand; (3) the mechanical properties (e.g., stiffness, elasticity) and structures (e.g., porosity size and density) of the hydrogels should match with the embedded tissue. For instance, hydrogels for repair of bone defects must possess excellent stiffness (up to MPa level) and adapted porous structure. While hydrogels for neural system should be soft (~1.5 kPa stiffness) and for muscle regeneration are expected to exhibit resistance to tension and compression; (4) multi-functionality of the hydrogels could be explored and achieve a synergetic effect. For instance, for tumor treatment, hydrogels being designed to possess both localized sustained-drug-releasing ability and photo-thermal ability showed greater advantage compared with mono-system. Moreover, thermo-responsive hydrogels displaying drug burst release under photo-irradiation could achieve a synergetic effect to inhibit tumor; (5) hydrogels with in vivo tracking ability could be further explored, and expand its biomedical usage. Continuous observation of hydrogels and the healing process is feasible if hydrogels could be detected by MRI, fluorescence, electric current, etc. [134, 135]; (6) hydrogels should be designed to fulfill complicated bio-functionality. For instance, in repair of myocardial ischemia, ideal hydrogels not only guide myocardium regeneration but promoted restoration of electrical conduction of the cardiac muscle. In full thickness cartilage defect, multi-phase hydrogels able to guide cartilage and subchondral bone repair respectively would be preferable.
In conclusion, self-healing hydrogels related to design, mechanism, characterization, and biomedical application were systematically reviewed here. Compared with conventional hydrogels, self-healing hydrogel exhibited greater advantages such as the prolonged lifetimes and improved mechanical performances derived from self-healing ability. Though some properties could be further explored, self-healing hydrogels already exhibit a promising future and may be extensively applicated in the biomedical area.
AcknowledgementThis work was supported by the National Natural Science Foundation of China (No. 31600778), and funded by Sichuan University (No. 2016SCU11044).
| [1] |
N.A. Peppas, J.Z. Hilt, A. Khademhosseini, R. Langer, Adv. Mater. 18(2006) 1345-1360. DOI:10.1002/(ISSN)1521-4095 |
| [2] |
W.E. Hennink, C.F. van Nostrum,Adv. Drug Deliv. Rev. 54(2002) 13. DOI:10.1016/S0169-409X(01)00240-X |
| [3] |
W. Shen, J. Luan, L. Cao, et al., Biomacromolecules 16(2015) 105-115. DOI:10.1021/bm501220a |
| [4] |
L. Cao, B. Cao, C. Lu, G. Wang, L. Yu, J. Ding, J. Ding,J. Mater. Chem. B 3(2014) 1268-1280. |
| [5] |
Y. Chen, Y. Li, W. Shen, et al., Sci. Rep. 6(2016) 31593. DOI:10.1038/srep31593 |
| [6] |
Y. Chen, J. Luan, W. Shen, et al.,ACS Appl. Mater. Interfaces 8(45) (2016) 30703.
|
| [7] |
B.V. Slaughter, S.S. Khurshid, O.Z. Fisher, A. Khademhosseini, N.A. Peppas, Adv. Mater. 21(2009) 3307-3329. DOI:10.1002/adma.v21:32/33 |
| [8] |
D.L. Taylor, I.H.P. Marc, Adv. Mater. 28(2016) 9060-9093. DOI:10.1002/adma.201601613 |
| [9] |
Z. Wei, J.H. Yang, J. Zhou, et al., Chem. Soc. Rev. 43(2014) 8114-8131. DOI:10.1039/C4CS00219A |
| [10] |
A. Phadke, C. Zhang, B. Arman, et al., Proc. Natl. Acad. Sci. 109(2012) 4383-4388. DOI:10.1073/pnas.1201122109 |
| [11] |
C.B. Highley, C.B. Rodell, J.A. Burdick, Adv. Mater. 27(2015) 5075. DOI:10.1002/adma.201501234 |
| [12] |
N. Huebsch, C.J. Kearney, X. Zhao, et al., Proc. Natl. Acad. Sci. 111(2014) 9762-9767. DOI:10.1073/pnas.1405469111 |
| [13] |
Z. Wei, J.H. Yang, X.J. Du, et al., Macromol. Rapid Commun. 34(2013) 1464-1470. DOI:10.1002/marc.v34.18 |
| [14] |
W.J. Yang, X. Tao, T. Zhao, et al., Polym. Chem. 6(2015) 7027-7035. DOI:10.1039/C5PY00936G |
| [15] |
F. Yu, X. Cao, J. Du, G. Wang, X. Chen, ACS Appl. Mater. Interfaces 7(2015) 24023-24031. DOI:10.1021/acsami.5b06896 |
| [16] |
V. Can, Z. Kochovski, V. Reiter, et al., Macromolecules 49(2016) 2281-2287. DOI:10.1021/acs.macromol.6b00156 |
| [17] |
E.A. Appel, M.W. Tibbitt, M.J. Webber, et al, Nat. Commun. 6(2015) 6295. DOI:10.1038/ncomms7295 |
| [18] |
F. Luo, T.L. Sun, T. Nakajima, et al., Macromolecules 47(2014) 6037-6046. DOI:10.1021/ma5009447 |
| [19] |
F. Liu, F. Li, G. Deng, et al., Macromolecules 45(2012) 1636-1645. DOI:10.1021/ma202461e |
| [20] |
D.C. Tuncaboylu, M. Sari, W. Oppermann, O. Okay, Macromolecules 44(2011) 4997-5005. DOI:10.1021/ma200579v |
| [21] |
D.C. Tuncaboylu, M. Sahin, A. Argun, W. Oppermann, O. Okay, Macromolecules 45(2012) 1991-2000. DOI:10.1021/ma202672y |
| [22] |
D.C. Tuncaboylu, A. Argun, M. Sahin, M. Sari, O. Okay, Polymer 53(2012) 5513-5522. DOI:10.1016/j.polymer.2012.10.015 |
| [23] |
H. Zhang, H. Xia, Y. Zhao, ACS Macro Lett. 1(2012) 1233-1236. DOI:10.1021/mz300451r |
| [24] |
B.K. Ahn, D.W. Lee, J.N. Israelachvili, J.H. Waite, Nat. Mater. 13(2014) 867-872. DOI:10.1038/nmat4037 |
| [25] |
M. Nakahata, Y. Takashima, H. Yamaguchi, A. Harada, Nat. Commun. 2(2011) 511. DOI:10.1038/ncomms1521 |
| [26] |
T. Kakuta, Y. Takashima, M. Nakahata, et al., Adv. Mater. 25(2013) 2849-2853. DOI:10.1002/adma.201205321 |
| [27] |
E.A. Appel, X.J. Loh, S.T. Jones, et al., J. Am. Chem. Soc. 134(2012) 11767-11773. DOI:10.1021/ja3044568 |
| [28] |
A.B. Ihsan, T.L. Sun, T. Kurokawa, et al., Macromolecules 49(2016) 4245-4252. DOI:10.1021/acs.macromol.6b00437 |
| [29] |
F. Luo, T.L. Sun, T. Nakajima, et al., Adv. Mater. 27(2015) 2722. DOI:10.1002/adma.v27.17 |
| [30] |
Y. Zhang, L. Tao, L. Shuxi, Y. Wei, Biomacromolecules 12(2011) 2894. DOI:10.1021/bm200423f |
| [31] |
S. Lü, C. Gao, X. Xu, et al., ACS Appl. Mater. Interfaces 7(2015) 13029. DOI:10.1021/acsami.5b03143 |
| [32] |
R. Chang, X. Wang, X. Li, H. An, J. Qin, ACS Appl. Mater. Interfaces 8(2016) 25544-25551. DOI:10.1021/acsami.6b08279 |
| [33] |
Z. Wei, J.H. Yang, Z.Q. Liu, et al., Adv. Funct. Mater. 25(2015) 1352-1359. DOI:10.1002/adfm.v25.9 |
| [34] |
G. Tachedjian, J.W. Mellors, H. Bazmi, J. Mills, Chem. Soc. Rev. 42(2013) 8106-8121. DOI:10.1039/c3cs60152h |
| [35] |
L. He, D.E. Fullenkamp, J.G. Rivera, P.B. Messersmith, Chem. Commun. 47(2011) 7497-7499. DOI:10.1039/c1cc11928a |
| [36] |
C.C. Deng, W.L.A. Brooks, K.A. Abboud, B.S. Sumerlin, ACS Macro Lett. 4(2015) 220-224. DOI:10.1021/acsmacrolett.5b00018 |
| [37] |
G. Deng, F. Li, H. Yu, et al., Acs Macro Lett. 1(2012) 275-279. DOI:10.1021/mz200195n |
| [38] |
S. Mukherjee, M.R. Hill, B.S. Sumerlin, Soft Matter 11(2015) 6152-6161. DOI:10.1039/C5SM00865D |
| [39] |
G. Li, J. Wu, B. Wang, et al., Biomacromolecules 16(2015) 3508-3518. DOI:10.1021/acs.biomac.5b01287 |
| [40] |
E.A. Appel, F. Biedermann, U. Rauwald, et al., J. Am. Chem. Soc. 132(2010) 14251-14260. DOI:10.1021/ja106362w |
| [41] |
M. Gosecki, B. Zgardzinska, M. Gosecka, et al., J. Phys. Chem. C 120(2016) 18323-18332. DOI:10.1021/acs.jpcc.6b06365 |
| [42] |
M. Shibayama, T. Tanaka, VolumePhase Transition and Related Phenomena of Polymer Gels, Responsive Gels:Volume Transitions I, Springer, Berlin, Heidelberg, 1993.
|
| [43] |
G. Akay, A. Hassanraeisi, D.C. Tuncaboylu, et al., Soft Matter 9(2013) 2254-2261. DOI:10.1039/c2sm27515e |
| [44] |
B. Ziółkowski, L. Florea, J. Theobald, F. Benitolopez, D. Diamond, Soft Matter 9(2013) 8754-8760. DOI:10.1039/c3sm51386f |
| [45] |
U. Gulyuz, O. Okay, Macromolecules 47(2014) 6889-6899. DOI:10.1021/ma5015116 |
| [46] |
M.P. Algi, O. Okay, Eur. Polym. J. 59(2014) 113-121. DOI:10.1016/j.eurpolymj.2014.07.022 |
| [47] |
T.T. Gao, N. Niu, Y.D. Liu, et al., RSC Adv. 6(2016) 43463-43469. DOI:10.1039/C6RA04271F |
| [48] |
B. Kurt, U. Gulyuz, D.D. Demir, O. Okay, Eur. Polym. J. 81(2016) 12-23. DOI:10.1016/j.eurpolymj.2016.05.019 |
| [49] |
C. Bilici, V. Can, U. Nöchel, et al., Macromolecules 49(2016) 7442-7449. DOI:10.1021/acs.macromol.6b01539 |
| [50] |
D.C. Tuncaboylu, A. Argun, M.P. Algi, O. Okay, Polymer 54(2013) 6381-6388. DOI:10.1016/j.polymer.2013.09.051 |
| [51] |
W. Li, H. An, Y. Tan, et al., Soft Matter 8(2012) 5078-5086. DOI:10.1039/c2sm07200a |
| [52] |
K. Xu, H. An, C. Lu, et al., Polymer 54(2013) 5665-5672. DOI:10.1016/j.polymer.2013.07.079 |
| [53] |
G. Li, Q. Yan, H. Xia, Y. Zhao,ACS Appl. Mater. Interfaces 7(2015) 12067-12073.
|
| [54] |
L. Guo, H. Zhang, D. Fortin, H. Xia, Y. Zhao, Langmuir 31(2015) 11709-11716. DOI:10.1021/acs.langmuir.5b03474 |
| [55] |
L. Yuan, L. Ren, X. Tian, et al., RSC Adv. 6(2016) 71425-71430. DOI:10.1039/C6RA14032G |
| [56] |
D. Shi, R. Liu, W. Dong, et al., RSC Adv. 5(2015) 82252-82258. DOI:10.1039/C5RA15991A |
| [57] |
L. Li, B. Yan, J. Yang, L. Chen, H. Zeng, Adv. Mater. 27(2015) 1294-1299. DOI:10.1002/adma.201405166 |
| [58] |
T.V. Chirila, H.L. Hui, M. Oddon, et al., J. Appl. Polym. Sci. 131(2014) 1001-1007. |
| [59] |
Jiaxi Cui., del Campo Aránzazu, Chem. Commun 48(2012) 9302-9304. DOI:10.1039/c2cc34701f |
| [60] |
G. Zhang, T. Ngai, Y. Deng, C. Wang, Macromol. Chem. Phys. 217(2016) 2172-2181. DOI:10.1002/macp.v217.19 |
| [61] |
Y. Lin, G. Li, J. Mater. Chem. B 2(2014) 6878-6885. DOI:10.1039/C4TB00862F |
| [62] |
Bastings M.C. Maartje, Koudstaal Stefan, R.E. Kieltyka, et al., Adv. Healthc. Mater. 3(2014) 70-78. DOI:10.1002/adhm.v3.1 |
| [63] |
L.Nicholas Fletcher, Lockett V. Christina, Dexter F. Annette, Soft Matter 7(2011) 10210-10218. DOI:10.1039/c1sm06261a |
| [64] |
X. Dai, Y. Zhang, L. Gao, et al., Adv. Mater. 27(2015) 3566. DOI:10.1002/adma.v27.23 |
| [65] |
C. Li, P. Chen, Y. Shao, et al., Small 11(2015) 1138-1143. DOI:10.1002/smll.v11.9-10 |
| [66] |
R. Du, J. Wu, L. Chen, et al., Small 10(2014) 1387-1393. DOI:10.1002/smll.v10.7 |
| [67] |
X. Qi, G. Ying, G. Chen, et al., RSC Adv. 5(2015) 41006-41012. DOI:10.1039/C5RA04115E |
| [68] |
S. Saha, J. Bachl, T. Kundu, D.D. Díaz, R. Banerjee, Chem. Commun. 50(2014) 3004-3006. DOI:10.1039/C3CC49869G |
| [69] |
S. Basak, J. Nanda, A. Banerjee, Chem. Commun. 50(2014) 2356-2359. DOI:10.1039/C3CC48896A |
| [70] |
M.L. Bender, M. Komiyama, CyclodextrinChemistry, Springer-Verlag, Berlin Heidelberg, 1978.
|
| [71] |
L. Peng, H. Zhang, A. Feng, et al., Polym. Chem. 6(2015) 3652-3659. DOI:10.1039/C5PY00296F |
| [72] |
C.B. Rodell, R.J. Wade, B.P. Purcell, N.N. Dusaj, J.A. Burdick, ACS Biomater. Sci. Eng. 1(2015) 277-286. DOI:10.1021/ab5001673 |
| [73] |
C.B. Rodell, N.N. Dusaj, C.B. Highley, J.A. Burdick, Adv. Mater. 28(2016) 8419. DOI:10.1002/adma.v28.38 |
| [74] |
L. Ouyang, C.B. Highley, C.B. Rodell, W. Sun, J.A. Burdick, ACS Biomater. Sci. Eng. 2(2016) 1743-1751. DOI:10.1021/acsbiomaterials.6b00158 |
| [75] |
K. Miyamae, M. Nakahata, Y. Takashima, A. Harada, Angew. Chem. Int. Ed. 127(2015) 8984-8987. |
| [76] |
Y.G. Jia, X.X. Zhu, Chem. Mater. 27(2015) 387-393. DOI:10.1021/cm5041584 |
| [77] |
T. Miao, S.L. Fenn, P.N. Charron, R.A. Oldinski, Biomacromolecules 16(2011) 3740. |
| [78] |
H. Chen, X. Ma, S. Wu, H. Tian, Angew. Chem. 53(2014) 14149-14152. DOI:10.1002/anie.v53.51 |
| [79] |
J.R. Mckee, E.A. Appel, J. Seitsonen, et al.,Adv. Funct. Mater. 24(18) (2014) 2706-2713.
|
| [80] |
M.J. Rowland, E.A. Appel, R.J. Coulston, O.A. Scherman, J. Mater. Chem. B 1(2013) 2904-2910. |
| [81] |
M. Nakahata, Y. Takashima, A. Harada, Macromol. Rapid Commun. 37(2015) 86-92. |
| [82] |
M. Tan, Y. Cui, A. Zhu, et al., Polym. Chem. 6(2015) 7543-7549. DOI:10.1039/C5PY01073J |
| [83] |
Y. Guo, X. Zhou, Q. Tang, et al., J. Mater. Chem. A 4(2016) 8769-8776. DOI:10.1039/C6TA01441K |
| [84] |
Z. Wei, J. He, T. Liang, et al., Polym. Chem. 4(2013) 4601-4605. DOI:10.1039/c3py00692a |
| [85] |
M. Zhong, X.Y. Liu, F.K. Shi, et al., Soft Matter 11(2015) 4235. DOI:10.1039/C5SM00493D |
| [86] |
P.S. Yavvari, A. Srivastava, J. Mater. Chem. B 3(2015) 899-910. DOI:10.1039/C4TB01307G |
| [87] |
S. Huang, L. Yang, M. Liu, et al., Langmuir 29(2013) 1238-1244. DOI:10.1021/la303855t |
| [88] |
T. Bai, S. Liu, F. Sun, et al., Biomaterials 35(2014) 3926-3933. DOI:10.1016/j.biomaterials.2014.01.077 |
| [89] |
T.L. Sun, F. Luo, T. Kurokawa, et al., Soft Matter 11(2015) 9355-9366. DOI:10.1039/C5SM01423A |
| [90] |
T.L. Sun, T. Kurokawa, S. Kuroda, et al., Nat. Mater. 12(2013) 932-937. DOI:10.1038/nmat3713 |
| [91] |
F. Luo, T.L. Sun, T. Nakajima, et al., Macromolecules 49(2016) 2750-2760. DOI:10.1021/acs.macromol.6b00235 |
| [92] |
B. Yang, Y. Zhang, X. Zhang, L. Tao, S. Li, Y. Wei, Polym. Chem. 3(2012) 3235-3238. DOI:10.1039/c2py20627g |
| [93] |
Y. Zhang, B. Yang, X. Zhang, et al., Chem. Commun. 48(2012) 9305-9307. DOI:10.1039/c2cc34745h |
| [94] |
W. Huang, Y. Wang, Y. Chen, et al., Adv. Healthc. Mater. 5(2016) 2813-2822. DOI:10.1002/adhm.v5.21 |
| [95] |
F. Ding, S. Wu, S. Wang, et al., Soft Matter 11(2015) 3971-3976. DOI:10.1039/C5SM00587F |
| [96] |
J. Ganguly, S. Maity, A. Datta, S. Lahiri, RSC Adv. 6(2016) 81060-81068. DOI:10.1039/C6RA15138H |
| [97] |
A.R. Karimi, A. Khodadadi, M. Hadizadeh, RSC Adv. 6(2016) 91445-91452. DOI:10.1039/C6RA17064A |
| [98] |
A.R. Karimi, A. Khodadadi, ACS Appl. Mater. Interfaces 8(2016) 27254-27263. DOI:10.1021/acsami.6b10375 |
| [99] |
X. Lu, Y.H. Ma, J. Yang, et al., Polym. Chem. 7(2016) 2037-2044. DOI:10.1039/C5PY01773D |
| [100] |
W.G. Skene, J.M. Lehn, Proc. Natl. Acad. Sci. 101(2004) 8270-8275. DOI:10.1073/pnas.0401885101 |
| [101] |
Maeda T., H. Otsuka, A Takahara, Prog. Polym. Sci 34(2009) 581-604. DOI:10.1016/j.progpolymsci.2009.03.001 |
| [102] |
G. Deng, C. Tang, F. Li, H. Jiang, Y. Chen, Macromolecules 43(2010) 1191-1194. DOI:10.1021/ma9022197 |
| [103] |
W. Yang, X. Wu, F. Liu, et al., RSC Adv. 6(2016) 34254-34260. DOI:10.1039/C6RA02366E |
| [104] |
M.C. Roberts, M.C. Hanson, A.P. Massey, E.A. Karren, P.F. Kiser, Adv. Mater. 19(2007) 2503-2507. DOI:10.1002/(ISSN)1521-4095 |
| [105] |
M. Vatankhahvarnoosfaderani, S. Hashmi, A. Ghavaminejad, F.J. Stadler, Polym. Chem. 5(2013) 512-523. |
| [106] |
Y. Dong, W. Wang, O. Veiseh, et al., Langmuir 32(2016) 8743-8747. DOI:10.1021/acs.langmuir.5b04755 |
| [107] |
V. Yesilyurt, M.J. Webber, E.A. Appel, Adv. Mater. 28(2015) 86-91. |
| [108] |
B. Wang, Y.S. Jeon, H.S. Park, J.H. Kim, Mater. Sci. Eng. C:Mater. Biol. Appl. 69(2016) 160-170. DOI:10.1016/j.msec.2016.06.065 |
| [109] |
H. Meng, P. Xiao, J. Gu, et al., Chem. Commun. 50(2014) 12277-12280. DOI:10.1039/C4CC04760E |
| [110] |
L. He, D. Szopinski, Wu Y. Yang, Gerrit A.Luinstra, Patrick Theato,ACS Macro Lett. 4(2015) 673-678. DOI:10.1021/acsmacrolett.5b00336 |
| [111] |
M.C. Kloetzel, The diels-alder reaction with maleic anhydride, Organic
|
| [112] |
H.L. Holmes, The Diels-Alder reaction ethylenic and acetylenic dienophiles, Organic Reactions, John Wiley & Sons, Inc., 2011.
|
| [113] |
X. Chen, M.A. Dam, K. Ono, et al., Science 295(2002) 1698-1702. DOI:10.1126/science.1065879 |
| [114] |
C.M. Nimmo, C.Shawn Owen, Shoichet S. Molly, Biomacromolecules 12(2011) 824-830. DOI:10.1021/bm101446k |
| [115] |
A. Kavitha, Singha K.Amalin Nikhil,ACS Appl. Mater. Interfaces 1(2009) 1427-1436. DOI:10.1021/am900124c |
| [116] |
S. Patai, Z. Rappoport, Thiol-disulfide Interchange. Sulphur-Containing Functional Groups, John Wiley & Sons, Inc., 2010, pp. 633-658.
|
| [117] |
Z.Q. Lei, H.P. Xiang, Y.J. Yuan, M.Z. Rong, M.Q. Zhang, Chem. Mater. 26(2014) 2038-2046. DOI:10.1021/cm4040616 |
| [118] |
P. Casuso, I. Odriozola, Pérez- V.A.San, et al.,Biomacromolecules 16(2015) 3552. |
| [119] |
A. Gantar, N. Drnovšek, P. Casuso, et al., RSC Adv. 6(2016) 69156-69166. DOI:10.1039/C6RA17327F |
| [120] |
J. Kalia, R.T. Raines, Angew. Chem. 120(2008) 7633-7636. DOI:10.1002/ange.v120:39 |
| [121] |
D. Seliktar, Science 336(2012) 1124-1128. DOI:10.1126/science.1214804 |
| [122] |
O. Okay, Self-healing hydrogelshormed via hydrophobic interactions:S. Seiffert (Ed.), Supramolecular Polymer Networks and Gels, Springer International Publishing, 2015, pp.101-142.
|
| [123] |
P. Wool Richard,Soft Matter 4(2008) 400-418.
|
| [124] |
Y. Qu, B.Y. Chu, J.R. Peng, et al., NPG Asia Mater. 7(2015). |
| [125] |
M.R. Nejadnik, X. Yang, M. Bongio, et al., Biomaterials 35(2014) 6918-6929. DOI:10.1016/j.biomaterials.2014.05.003 |
| [126] |
T.C. Tseng, L. Tao, F.Y. Hsieh, et al., Adv. Mater. 27(2015) 3518-3524. DOI:10.1002/adma.v27.23 |
| [127] |
L. Han, X. Lu, M. Wang, et al., Small 13(2017) 1601916. DOI:10.1002/smll.v13.2 |
| [128] |
R. Dong, X. Zhao, B. Guo, P.X. Ma, ACS Appl. Mater. Interfaces 8(2016) 17138-17150. DOI:10.1021/acsami.6b04911 |
| [129] |
C. Zhu, J. Zhao, K. Kempe, P. Wilson, et al., Macromol. Biosci. 17(2017) 1600320. DOI:10.1002/mabi.201600320 |
| [130] |
L. Ruan, H. Zhang, H. Luo, et al., Proc. Natl. Acad. Sci. 106(2009) 5105-5110. DOI:10.1073/pnas.0900026106 |
| [131] |
Q. Liu, C. Zhan, A. Barhoumi, et al., Adv. Mater. 28(2016) 6680-6686. DOI:10.1002/adma.201601147 |
| [132] |
H. Lu, Y. Zhang, L. Xiong, et al., ACS Appl. Mater. Interfaces 8(2016) 29088-29100. DOI:10.1021/acsami.6b11043 |
| [133] |
J. Liu, K. Wang, J. Luan, Z. Wen, L. Wang, Z. Liu, et al., J. Mater. Chem. B 4(2016) 1343-1353. DOI:10.1039/C5TB02459E |
| [134] |
K. Lei, Q. Ma, L. Yu, J. Ding, J. Mater. Chem. B 4(2016) 7793-7812. DOI:10.1039/C6TB02019D |
| [135] |
K. Lei, W. Shen, L. Cao, L. Yu, J. Ding, Chem. Commun. 51(2015) 6080-6083. DOI:10.1039/C5CC00049A |