 2017, Vol. 28
2017, Vol. 28 



b Department of Gastroenterology, Hospital(T. C. M) Affiliated to Southwest Medical University, Southwest Medical University, Luzhou 646000, China
Quantum dots (QDs) display special electronic and optical properties that are tunable with different sizes and shapes [1, 2]. QDs can be produced by means of physical, chemical, and biological approaches [3]. The physical synthesis methods, such as laser physical vapor deposition and laser irradiation of large particles, are typically facile, time and cost-saving. The chemical synthesis routes, including the water-phase and organic-phase synthesis methods, are the classical approaches to prepare QDs with monodispersity and good optical performance [3]. Significant progress has been made in controlling the size and shape, enhancing the stability and elucidating the mechanisms of QD formation [3]. The biosynthesis methods are a green synthesis route and provide an alternative that not only carry out the synthesis reaction in mild conditions, but also endue the QDs with inherent biostability and biocompatibility [3]. Similar to the chemically synthesized QDs, the biosynthesized QDs exhibit sizetunable emission, simultaneous excitation of different-sized QDs by a single light source, and component-tunable broad spectrum windows [3]. QDs emit narrow symmetric bands under a wide excitation range, possess anti-photobleaching stability, and exhibit advanced molecular imaging possibilities as well as ultrasensitive bioassays and diagnostics in vitro and in vivo [4, 5]. In addition, biofunctionalized QDs facilitate modification for widely biomedical application (Fig. 1) [6-9]. Therefore, QDs with unique optical properties become very attractive vectors for imaging-guided therapy [10-12].

|
Download:
|
| Fig. 1. Schematic diagram of typically bio-functionalized core/shell QDs. A, B, C and D are carboxyl, thiol, amino and streptavidin coated core/shell QDs, respectively. | |
{kind=link}
Small-interfering RNA (siRNA)-based therapeutics hold great potential to target a large part of the currently undruggable genes and generate entirely new therapeutic paradigms in diseases, including cancer, infectious diseases, degenerative diseases, and cardiovascular diseases, among others [13-17]. However, it is difficult for naked siRNAs to enter the cells directly because of their negative charges. Overcoming the lipid bilayer to deliver siRNA into cells has remained a major challenge to solve for widespread development of RNA therapeutics [18, 19]. This weakness limits the application of siRNAs in the treatment of diseases, and therefore it is necessary to develop siRNA delivery systems for RNAi therapy [20-22].
QDs can potentially promote the efficient delivery of siRNAs into target cells and track the distribution of siRNA in cells in vitro or in tissues in vivo [23]. Many researches have shown great interest in using QDs as an effective theranostic agent to deliver siRNA and track individual molecules [24, 25]. To date, several strategies have been reported for QD/siRNA delivery, including coencapsulating QDs and siRNA into cationic gene vectors to create QD/siRNA nanocomplexes, coupling functionalized QDs with cationic materials directly as a core/shell conjugation for siRNA theranostic delivery and grafting streptavidin-QDs with biotin-siRNA based on the multivalent binding capability of streptavidin to biotin [26]. The sizes of QDs are significantly increased and the emission spectra of QDs may be affected after binding with siRNA to forming QDs/siRNA complexes. In addition, the cytotoxicity of QDs is usually decreased by complexing with siRNA. Herein, we review theranostic QD/siRNA assembles for enhancing delivery of siRNA and facilitating evaluation of therapeutic efficacy via imaging of QDs, with special attention to carbonaceous quantum dots for delivery of siRNA.
2. Intracellular uptake and release mechanisms of siRNA/QD nanoassemblesThe electrostatic complexation of cationic QDs with anionic siRNA showed effective siRNA binding and intracellular delivery. Meanwhile, QDs were employed to illuminate the uptake and release mechanism of siRNA complexes. QD-mediated fluorescence resonance energy transfer revealed that lipopexes delivered via a nanochannel electroporation could directly release siRNA in the cytoplasm without going through the endocytosis route, which unraveled the responsible mechanism for efficient gene delivery (Fig. 2) [27]. Cationic polyethylene imine (PEI), poly-ε-caprolactone (PCL), and poly(ethylene glycol) (PEG) triblock copolymer (PEG-PCL-PEI) was designed for delivery of siRNA. A fluorescence resonance energy transfer pair composed of AF647-congjugated siRNA against GAPDH and QD was incorporated into PEG-PCL-PEI for illuminating the mechanism of siRNA-unpacking and evaluating the knockdown efficiency in vitro and in vivo. The observation of fluorescence resonance energy transfer demonstrated that AF647-siRNA was dissociated from PEG-PCL-PEI/AF647-siRNA/QD complexes stimulated by heparin. PEG500-PCL10000-PEI2500/ siGAPDH/QD complexes revealed good transfection efficiency (61% ± 5% knockdown in vitro, 55% ± 18% knockdown in vivo), and poor performance was found for their hydrophilic counterparts (PEG5000-PCL10000-PEI2500) (13% ± 6% knockdown in vitro, 30% ± 17% knockdown in vivo). Therefore, this theranostic system has a potential for illuminating basic principles of the delivery process and enlightening a more rational design of amphiphilic gene carriers [28].

|
Download:
|
| Fig. 2. Schematic diagram of intracellular uptake and release mechanism of siRNA/ QD nanoassembles. | |
{kind=link}
3. QD/siRNA delivery via the electrostatic complexation
The CdSe/CdS/ZnS QDs can be encapsulated into the hydrophobic pockets of amphiphilic PEI derivative polymer composites (QDPEI), while their polycationic outer surfaces can bind with siRNA and efficiently interact with Mardin-Darby canine kidney (MDCK) cell line. For example, QD-PEI/siGFP was effectively internalized according to the red fluorescence intensity of QD-PEI and almost completely silenced GFP gene expression in MDCK-GFP cells. Therefore, QD-PEI/siRNA was an effective vector for siRNA delivery [29]. CdSSe/ZnS QDs-PEI delivered siRNA into two glioblastoma cell lines (U87 and U251) to target human telomerase reverse transcriptase (TERT) gene. The gene expression level (50% for U87, 43% for U251) and protein expression level (51% for U87, 50% for U251) of TERT were observed to decrease substantially after transfecting the tumor cells for 48 h. Meanwhile, the silencing of TERT gene suppressed the proliferation of glioblastoma cells. In addition, no obvious cytotoxicity from these QD-PEI nanoplexes was observed over at 10 times of the transfected doses. Therefore, this QDs-PEI could be used as a safe and efficient gene nanocarrier for siRNA delivery [30]. A kind of arginine-modified CdSe/ZnSe QDs were prepared as cationic nanocarriers and showed low cytotoxicity and efficient knockdown effect of HPV18 E6 gene in the HeLa cells [31]. QDs with a small size facilitate delivery of siRNA into tumor tissues via the enhanced permeability and retention (EPR) effect. To achieve a pH-responsive hybrid QDs (QD-DG-9R/siRNA) for targeting hypoxic tumor siRNA delivery, CdTe QDs were coated by a functional shell composed of 2-deoxyglucose (DG)-PEG connected with the compound of lipoic acid, lysine and 9-poly-Darginine (LA-Lys-9R) by a hydrazone bond. DG was recognized and transported into the cells by GLUT1 on the cell membrane, and 9R as a gene carrier was employed to deliver siRNA against HIF-1a to the cytoplasm for the treatment of hypoxic tumors. Importantly, the hydrazone bond avoided GLUT1 receptor-mediated endocytic recycling and irreversibly delivered siHIF-1α enhanced hypoxic tumor targeting. QD-DG-9R/siHIF-1α suppressed ~60% HIF-1α at mRNA level in HepG2 cells and then inhibited the proliferation of HepG2 cells under the hypoxic condition. Moreover, QD-DG-9R/ siHIF-1α repressed the subcutaneous tumor growth in a HepG2 xenograft model and showed few side effects and little toxicity. Consequently, the hypoxic tumor targeting and pH-response QD/ siRNA complexes have great potential for targeted gene therapy of hypoxic tumors [32]. Zhao et al. fabricated cationically-modified CdSe QDs by coating a layer of the amine functionalized PEG on the surface of carboxyl-terminated QDs. The QD solution was mixed with Survivin siRNA to form QD-siSurvivin complexes via electrostatic complexation. Oral squamous cell carcinoma Tca8113 cells were successfully transfected by QDs-siSurvivin complexes, and the red fluorescence from QDs and green fluorescein amidite fluorescence from siRNA could both be easily observed after 6 h of incubation. siRNA was released into the cytoplasm and Survivin mRNA level was reduced [33]. A palmitoylated peptide (JB577) was synthesized to coat the outer shell of CdSe/ZnS QD via the histidine residues of JB577 and bind siRNA via the positively charged KIKK sequence of JB577, and then QD-JB577/siRNA complexes were obtained. By attaching siRNA for luciferase to QD-JB577, 70% inhibition of mRNA was observed after the transfection for 24-48 h in a cell line over-expressing luciferase. Moreover, the knockdown efficiencies of lysosomal acid sphingomyelinase (ASMase) or neutral sphingomyelinase 2 (NSMase2) were 45% or 41% after treatment by QD-JB577/ siASMase or QD-JB577/siNSMase2. In addition, there was no observed toxicity in contrast to the toxicity observed with lipofectamines. Therefore, QD-JB577/siRNA complexes provide a promising strategy for therapeutic siRNA delivery and potential useful for treating variety of diseases in the future [34]. The Mn: ZnSe QD was modified by cationic polyelectrolyte PAH to generate positive surface potential for complexing with K-Ras siRNA molecules (QD/PAH/siK-Ras). QD/PAH/siK-Ras was further incubated with PAH to form QD/PAH/siK-Ras/PAH that was eventually functionalized with folic acid to obtain folic acid-conjugated nanoplexes (QD/PAH/siK-Ras/PAH-folic acid). The ligand-modified QDs/siRNA complexes increased the uptake of siRNA by specific interaction between the ligand and receptor in tumor cells, which could enhance the efficiency of gene disruption of siRNA. The nanoplexes transfected over 70% of Panc-1 cells and suppressed 60% of K-Ras gene at mRNA level. The folic acid-conjugated nanoplexes with high transfection efficiency and gene disruption should serve as a promising candidate for biomedical applications [35]. The Mn:ZnSe/ZnS/ZnMnS QD cross-linked by 2-aminoethanethiol was mixed with siRNA against the mutant K-Ras gene to form QD/siRNA complexes, which were encapsulated into a folate-modified liposome to prepare QD/siRNA-liposome hybrid nanoparticles. The folate-modified QD/siRNA-liposome hybrid nanoparticles significantly enhanced the transfection efficiency to around 70% compared to 9% of the hybrid nanoparticles without folate modification. More importantly, the folate-modified QD/siKRas-liposome hybrid nanoparticles suppressed 54% Panc-1 cells by mRNA assay, and lower cell viability were observed in the hybrid nanoparticles group because the K-Ras gene was silenced. The QD/ siRNA-liposome hybrid nanoparticles may be used to targeted therapy for cancer with folate receptor [36].
4. QD/siRNA delivery via the interaction between streptavidin and biotinStreptavidin is composed of four subunits and each of the four subunits can bind one biotin molecule with a very high affinity (Kd = 10-15 mol/L). Consequently, QDs were utilized to deliver siRNAs dependent on the interaction between streptavidin and biotin. Cell-derived microparticles were modified by adding DSPEPEG-biotin to the starved cells for releasing biotinylated vesicles, which were electroporated with siRNA and then labeled by incubating with streptavidin-conjugated QD for producing QDlabeled siRNA vesicles. The QD-labeled siVEGF vesicles diffused toward the whole site by detecting the fluorescence signals of QD and exhibited tumor-inhibitory effects in the model of A2058 human melanoma xenografts in vivo via intratumoral injection. Therefore, the cell-derived microparticles can be developed as theronostic vectors for imaging and siRNA delivery [37]. QD coupled with streptavidin was modified by biotin-ligands [GE11 for EGFR and c(RGDfK) for αvb3] and biotin-aminocaproic acidsiSurvivin for obtaining dual receptor-targeting gene carrier (dualtargeted QD-siRNA). The dual-targeted QD-siRNA increased cellular uptake efficiency by quantitative flow cytometry and inductively coupled plasma mass spectrometry (ICP-MS), and achieved higher gene silencing efficiency than lipofectamine 2000/siRNA complexes [38]. This targeted QD-siRNA nanocarrier based on streptavidin-biotin interaction might be a new theranostic carrier for siRNA. Based on the multivalent binding capability of streptavidin to biotin, CdSe/ZnS QDs were decorated with multiple copies of targeting ligands (p160 peptide) and siRNA against folate receptor a. Confocal imaging and quantitative flow cytometry showed that QD-(siRNA + p160) was able to specifically target human breast cancer MCF-7 cells via receptor-mediated endocytic pathway. Western blot and quantitative ELISA analysis indicated the expression level of folate receptor α had a significant decrease at 50 nmol/L. The nanocarriers not only achieved gene silence in a cell-specific manner but also achieved real-time tracking during siRNA intracellular delivery [39].
5. QD/siRNA delivery via the chemical conjugationWu et al. developed functional CdSe/ZnS QD complexes (QDSMCC/siSOX9) by sulfosuccinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC) activation of PEG-coated QDs as the gene vector of siRNA to study the effect of SOX9 RNA interference on the chondrogenic differentiation of mesenchymal stem cells (MSCs). QD-SMCC effectively bound and delivered siRNA into the MSCs, followed by efficient siRNA escape from the endosomes. Consequently, the transfection efficiency of QD-SMCC (68.6%) was superior to that of PEI (56.4%). The siSOX9 released from QD-SMCC/siSOX9 retained its structural integrity and effectively inhibited the targeted gene expression, leading to reduced chondrogenic differentiation of MSCs and delayed cartilage repair. Because the ZnS shell and PEG coating layer greatly reduced the cytotoxicity of the QD-SMCC, QDs were excreted from living cells instead of dead cells. The results indicate that SOX9 is imperative for the chondrogenesis of MSCs and QDSMCC has great potential for therapeutic siRNA delivery and realtime tracking of transfection [40]. Amino-modified QDs were conjugated to thiol-containing siRNA and F3 tumor-targeting peptides using sulfosuccinimidyl 6(3'-[2-pyridyldithio]-propionamido) hexanoate and sulfo-SMCC cross-linkers. Approximate 20 F3 peptides and 1 siRNA duplex were coupled to one QD particle (F3-QD-siRNA), and the loaded siRNA did not affect the function of F3 peptide. After delivery to cells and release from their endosomal entrapment, a 29% reduction in EGFP was observed in EGFPexpressing HeLa cells treated by F3-QD-siRNA. By designing the siRNA sequence against a therapeutic target (e.g., oncogene) instead of EGFP, the technology explored in this study may be adapted to treat and image diseases such as cancer [41]. Subramaniam et al. reported a rapid sonochemical synthetic methodology for generating a library of highly biocompatible ZnSAgInS(2) (ZAIS) QDs for cellular imaging and siRNA delivery. The physicochemical properties of ZAIS QDs could be tuned over the entire visible spectrum by varying the chemical composition of the precursors. PEI was conjugated to ZAIS QDs via 3-mercaptopropionic acid to synthesize QD-PEI which bound with siRNA to form QD-PEI/siRNA complexes. The QD-PEI/siEGFP complexes showed efficient target gene silencing effects (80% decrease of the green fluorescence after 3 days of transfection) as evidenced by the ntracellular red fluorescence of the QDs in brain cancer U87-EGFP cells with negligible cytotoxicity, thereby allowing ZAIS QDs to be applied for molecular imaging and effective delivery of siRNA [25, 42]. Singh et al. performed a systematic study to evaluate siRNA coupling strategies using an amino-PEGylated QDs system loaded with two kinds of siRNA macromolecules per nanoparticle. It was found that the target gene (GFP-Ago2/Luc-CXCR4) in HeLa cells was efficiently knocked down by the labile formulation (90%). Due to the susceptible cross-linkers which was cleaved in the reducing intracellular environment, then conjugated siRNAs were released from the nanoparticle, subsequently, siRNAs became available to complex with the RISC for efficient silencing effect. In addition, the nanoparticle with a longer tether might enhance the macromolecule flexibility and improve accessibility of siRNA [43].
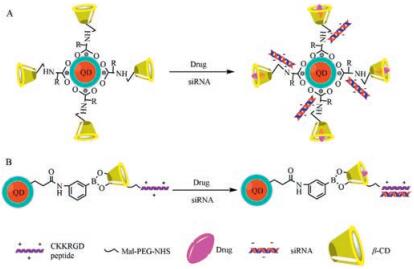
6. Co-delivery of siRNA and drug via β-CD modified QDL-Arginine as a linker was used to conjugate CdSe/ZnSe QD and hydropropyl-cyclodextrins (HP-CDs, including HP-α-CD, HP-β-CD, HP-γ-CD) to form HP-CD-QDs nanocarriers for siRNA (targeting Bcl-2 gene, siBcl-2) or/and antitumor drugs (carboplatin, paclitaxel and doxorubicin) delivery. HP-CDs-L-arginine modification did not only alter the optical properties of QDs but also reduce the cytotoxicity of QDs. HP-CD-QD/siRNA complexes were prepared at an siRNA:QD molar ratio of 1:1, and the complexes efficiently delivered siBcl-2 into A549 cells and offered a better siRNA transfection in A549 cells compared to siPort NeoFX (an siRNA transfection agent). mRNA expression levels of the Bcl-2 gene were ranged from 33% to 40% after the transfection of HP-CD-QD/siBcl-2 complexes. When the complexes loading one or two anticancer drug(s) for siBcl-2 delivery were used to transfect into A549 cells, over 80% Bcl-2 gene was suppressed and the cytotoxicity was increased by three-fold to four-fold compared with free drug treatments. Therefore, the multifunctional HP-CD-QD nanocarriers can effectively deliver siRNA and different antitumor drugs simultaneously into lung cancer cells and enhance the synergistic effect of combination therapy (gene therapy and chemotherapy) regimens for lung cancer therapy (Fig. 3A) [20]. Similarly, CdSe/ ZnSe QD modified with β-CD coupled to L-histidine could simultaneously deliver siMDR1 and doxorubicin to reverse the multidrug resistance of HeLa cells [44]. A carboxylated CdTe/ZnS QD was conjugated with amino-beta-cyclodextrin (NH2-b-CD) via a 3-aminophenyl boronic acid linker to produce NH2-b-CD-QD, which was further coupled with a RGD peptide (CKKRGD) by a maleimidep-(PEG)4-N-hydroxysuccinimide linker to obtain RGDb-CD-QD nanocarrier. The RGD peptide in the nanocarrier bound siRNA against peroxisome proliferator-activated receptor gamma (PPAR-γ, siPPAR-γ) by electrostatic interaction, and dexamethasone was incorporated into the hydrophobic cavity of β-CDs in the nanocarrier to prepare a co-delivery system of siRNA and dexamethasone by RGD-β-CD-QD (Fig. 3B). The RGD peptide not only facilitated the complexation of siPPAR-g and delivered siRNA into human mesenchymal stem cells but also leaded to cellular uptake of nanoparticles by RGD receptor. Co-delivery of dexamethasone and siPPAR-γ by RGD-β-CD-QD nanocarrier significantly expedited and enhanced the osteogenesis differentiation of human mesenchymal stem cells in vitro and in vivo by combined effect of small molecule and RNAi. Furthermore, the stable fluorescent signal of RGD-β-CD-QDs nanocarrier enabled realtime monitoring of the nanocarrier/drugs cellular uptake in vitro and long-term tracking (3 weeks) of human mesenchymal stem cells after implantation in vivo. Therefore, the RGD-β-CD-QDs nanocarrier provides a powerful tool to simultaneously enhance differentiation and long-term tracking of human mesenchymal stem cells in vitro and in vivo for regenerative medicine [45].

|
Download:
|
| Fig. 3. Schematic diagram of siRNA adsoption and drug loading onto b-CD modified QD. A: Co-delivery of dexamethasone and siRNA via RGD peptide modified b-CD-QD conjugation; B: Co-delivery of doxorubicin and siRNA via L-amino acid modified b-CD-QD conjugation. | |
{kind=link}
The clinical application of QDs has been being questioned due to their potential and inherent cytotoxicity. Most of semiconductor-based QDs contain highly toxic elements such as Cd or Te. Taken commonly CdSe QDs as an example, the Cd2+ ions would eventually release into the cytoplasm during the biodegradation process in the cell. Next, the ions would bind to the sulfohydryl groups of mitochondria proteins and then inhibit mitochondrial respiration. Subsequently, the inherent cytotoxicity caused by ions would occur [46]. A recent result revealed a clear correlation between the toxicity and degradation of core-only CdTe QD in vitro and in vivo. Typically, the CdTe QDs efficiently inhibited the activation of IKKα and IKKβ, resulting in the suppression of both the canonical and the non-canonical nuclear factor-kB signaling pathways [47]. Furthermore, inhibition of nuclear factor-kB downregulated anti-apoptotic genes and promoted apoptosis in cancer cells [48]. Interestingly, this core-only CdTe QD as an anticancer agent was utilized to treat cancer in vivo and displayed a desired anticancer effect due to the toxicity of CdTe QDs [47]. Coating with biocompatible materials on QDs could reduce the toxicity, but the inherent risks of QDs still existed [25, 49]. Chitosan coated CdSe QDs and PEGylated CdSe/ZnS QDs exhibited significant toxicity in some studies [50, 51]. In response to the above issues, non-toxic elements QDs have been developed for siRNA delivery in the recent years.
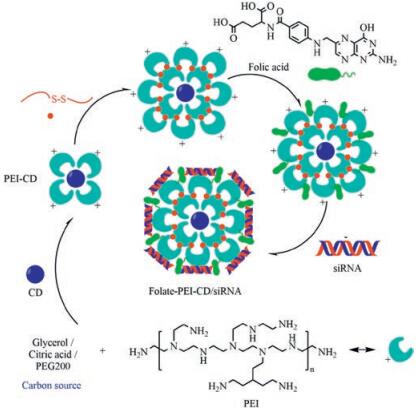
7. Carbon dots delivery of siRNACarbonaceous quantum dots or carbon quantum dots known as carbon dots (CDs) have emerged as an alternative to traditional semiconductor-based QDs as non-metallic fluorescent probes [52-54]. CDs surface passivation by amino compounds as required for obtaining exacerbated intrinsic fluorescence properties allows direct installation of cationic charge density at the surface of the nanoparticles. CDs have been explored as non-viral vectors for nucleic acid by chemical modification on purpose [55]. Cationic CDs facilitate to bind plasmid DNA and siRNA with anionic charges for gene delivery. Kim et al. reported on a ternary complex composed of PEI-functionalized CDs, PEI-functionalized gold nanoparticles and plasmid DNA for in vitro transfection of cancer cells and real-time monitoring of plasmid cellular trafficking [56]. Branched PEI-based CDs were prepared from branched PEI by oxidation and a modified hydrothermal reaction. The resulting cationic CDs proved efficient for in vitro transfection of MCF-7 cells using EGFP as a reporter gene [57]. Alkyl-PEI was used to react with CDs for surface passivation to synthesize cationic alkyl-PEI-CDs, which allowed for effective siRNA binding via electrostatic interaction to form alkyl-PEI-CDs/siRNA complexes. The formed CDs/siRNA complexes led to highly efficient gene delivery in vitro and 65% firefly luciferase gene of Luc-4T1 cells were specifically inhibited by firefly luciferase targeted siRNA (siLuc). Furthermore, the CD/siLuc compexes were directly injected into the Luc-4T1 tumor tissues and relative level of luciferase expression reduced to 62% after the treatment. Meanwhile, the CD/siLuc compexes maintained the fluorescence properties and high biocompatibility in vitro and in vivo. The work of Wang et al. highlighted the promise of alkyl-PEI-CD as a novel imaging-trackable nanocarrier for efficient gene delivery and optical molecular imaging [58]. A cationic CD was synthesized with PEG200 and PEI by a microwaveassisted method. The PEI-CDs bound siRNA efficiently when CDs were mixed with siRNA at a 20:1 ratio. AaSRC and AaSNF7 genes of Aedes aegypti larvae were targeted by PEI-CD/siAaSRC and PEI-CD/ siAaSNF7 through oral route. The efficient siRNA delivery via PEICDs caused mortality of mosquito larvae. Due to the fluorescent property of CDs, distribution of siRNA within the larval body could easily be monitored. A few PEI-CD/siRNA complexes were detected in the tissues after 24 h of exposure through food. Presence of complexes within larval tissues gradually increased with increasing time. Finally, knockdown of AaSRC and AaSNF7 genes in mosquito larvae by feeding PEI-CD/siAaSRC and PEI-CDs/siAaSNF7 caused high mortality. Therefore, this technology has the potential to become a sustainable, targeted, and eco-friendly insect management method [59]. A new cationic CD was fabricated by pyrolysis of citric acid and branched PEI under microwave radiation. The cationic CDs bound with firefly luciferase gene targeted siRNA (siLuc) for preparing CDs/siLuc complexes, which were rapidly internalized by A549 cells and monitored with a confocal laser scanning fluorescence microscopy. In Luc-A549 cells, the CD/siLuc complexes caused a 55% specific gene silencing at a CD/siLuc weight ratio of 12, and an 85% gene knockdown for weight ratios in the range 50-100. However, it needs to be further improved the in vivo transfection efficiency and biocompatibility of this cationic CDs produced by microwave radiation [60]. PEIadsorbed CDs were utilized to deliver siRNA against Survivin gene (siSurvivin) into human cancer cell line MGC-803. This PEI-CD nanocarrier exhibited excellent biocompatibility and a significant increase in cellular delivery of siSurvivin, inducing efficient knockdown for Survivin protein to 6.1%. The CDs-based and PEIadsorbed complexes both as imaging agents and siRNA nanocarriers could be applied for a broad range of siRNA delivery systems for cancer therapy [55]. Folate-conjugated reducible PEI passivated CDs (folate-rPEI-CD) could function as a siRNA carrier and release siRNA in reductive environment as shown in electrophoresis. Folate-rPEI-CDs encapsulating multiple siRNAs (EGFR and cyclin B1) (folate-rPEI-CDs/pooled siRNA) reduced the survival of H460 lung cancer cells to 30% (Fig. 4). The mRNA gene expressions of cyclin B1 and EGFR were both suppressed lower than one fold in folate-rPEI-CDs/pooled siRNA group from 12 h to 48 h. In addition, folate-rPEI-CDs/pooled siRNA sustained gene silencing ability for 48 h. Furthermore, folate-rPEI-CDs/pooled siRNA nanoagents specifically accumulated at lung region and continuously inhibited the growth of H460 tumor by 50% according to bioluminescent imaging for at least 2 weeks via aerosol delivery. Therefore, the novel theranostic folate-rPEI-CDs/siRNA nanoagents have a potential in lung cancer targeting and treatment [61].

|
Download:
|
| Fig. 4. Schematic diagram of synthesis route of Folate-PEI-CDs/siRNA nanoassembles. | |
{kind=link}
8. Conclusions and outlook
QDs/siRNA nanoassembles can be fabricated by the coencapsulation of QDs, siRNAs and biomaterials, the electrostatic complexation of QDs-cationic materials conjugation and siRNAs, or the streptavidin-biotin interaction between the modified QDs and siRNAs. Bio-functionalized QDs/CDs cannot only deliver the therapeutic siRNAs into cells in vitro and target tissues in vivo, but also track the trans-membrane process in vitro or distribution in vivo of the siRNAs. In addition, the release of siRNA from QDs/ siRNA nanoassembles can be monitored and imaged easily. As a promising theranostic platform, QDs/siRNA nanoassembles have been used to disturb plenty of genes for treatments of various diseases. However, due to the inherent toxicity of semiconductorbased QDs, they need to be improved for further clinical applications [62, 63]. Carbonaceous QDs with good biocompatibility may be an approach to eliminate the inherent toxicity of semiconductor-based QDs [52, 64]. It has been demonstrated PEImodified CDs can bind with siRNAs to form CD/siRNAs nanoassembles, which can deliver and track siRNAs efficiently into cells. In the near future, semiconductor-based QDs will be further modified for safe and stable QDs/siRNA nanoassembles, which can be used to illuminate the biodistribution of nanoformulations and the therapeutic mechanism of siRNAs. Carbonaceous QDs will be developed for the potential clinical application, and new QDs/ siRNAs nanoassembles will be studied for the treatment of various diseases.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 81602699), the National High Technology Research and Development Program of China (No. 2015AA020309), and the China Postdoctoral Science Foundation funded project (No. 2015M570791).
| [1] |
A.P. Alivisatos, Science 271(1996) 933-937. DOI:10.1126/science.271.5251.933 |
| [2] |
X. Dai, Y. Deng, X. Peng, Y. Jin, Adv. Mater.(2017). DOI:10.1002/adma.201607022 |
| [3] |
J. Zhou, Y. Yang, C.Y. Zhang, Chem. Rev. 115(2015) 11669-11717. DOI:10.1021/acs.chemrev.5b00049 |
| [4] |
X. Gao, Y. Cui, R.M. Levenson, L.W. Chung, S. Nie, Nat. Biotechnol. 22(2004) 969-976. DOI:10.1038/nbt994 |
| [5] |
Y. Zhang, T.H. Wang, Theranostics 2(2012) 631-654. DOI:10.7150/thno.4308 |
| [6] |
R. Bilan, I. Nabiev, A. Sukhanova, Chembiochem 17(2016) 2103-2114. DOI:10.1002/cbic.v17.22 |
| [7] |
A. Mukherjee, Y. Shim, Myong J.Song, Biotechnol. J. 11(2016) 31-42. |
| [8] |
S.L. Liu, Z.G. Wang, Z.L. Zhang, D.W. Pang, Chem. Soc. Rev. 45(2016) 1211-1224. DOI:10.1039/C5CS00657K |
| [9] |
C.R. Kagan, C.B. Murray, Nat. Biotechnol. 10(2015) 1013-1026. |
| [10] |
D. Radenkovic, H. Kobayashi, E.Semmelweis Remsey-, A.M. Seifalian, Nanomedicine 12(2016) 1581-1592. DOI:10.1016/j.nano.2016.02.014 |
| [11] |
E. Cassette, M. Helle, L. Bezdetnaya, et al., Adv. Drug Deliv. Rev. 65(2013) 719-731. DOI:10.1016/j.addr.2012.08.016 |
| [12] |
A.M. Pekkanen, M.R. DeWitt, M.N. Rylander, J. Biomed. Nanotechnol 10(2014) 1677-1712. DOI:10.1166/jbn.2014.1988 |
| [13] |
X. Chi, P. Gatti, T. Papoian, Drug Discov. Today 22(2017) 823-833. DOI:10.1016/j.drudis.2017.01.013 |
| [14] |
F. Mottaghitalab, A. Rastegari, M. Farokhi, et al., Int. J. Pharm. 524(2017) 312-329. DOI:10.1016/j.ijpharm.2017.03.092 |
| [15] |
P. Ofek, G. Tiram, Satchi-R.Fainaro, Adv. Drug Deliv. Rev.(2017). DOI:10.1016/j.addr.2017.01.008 |
| [16] |
D. Scherman, A. Rousseau, P. Bigey, V. Escriou, Gene Ther. 24(2017) 151-156. DOI:10.1038/gt.2017.6 |
| [17] |
R.Almeida Titze-de-, C. David, Titze-de-S.S.Almeida, Pharm. Res. 34(2017) 1339-1363. |
| [18] |
P.R. Cullis, M.J. Hope, Mol. Ther.(2017). DOI:10.1016/j.ymthe.2017.03.013 |
| [19] |
C. Selvam, D. Mutisya, S. Prakash, K. Ranganna, R. Thilagavathi, Chem. Biol. Drug Des.(2017). DOI:10.1111/cbdd.12993 |
| [20] |
J. Li, Y. Wang, S. Xue, et al., Int. J. Nanomed. 11(2016) 4609-4624. DOI:10.2147/IJN |
| [21] |
S.F. Dowdy, Nat. Biotechnol. 35(2017) 222-229. DOI:10.1038/nbt.3802 |
| [22] |
Y. Huang, Mol. Ther. Nucleic Acids 6(2017) 116-132. DOI:10.1016/j.omtn.2016.12.003 |
| [23] |
K.D. Wegner, N. Hildebrandt, Chem. Soc. Rev. 44(2015) 4792-4834. DOI:10.1039/C4CS00532E |
| [24] |
J.M. Knipe, J.T. Peters, N.A. Peppas, Nano Today 8(2013) 21-38. DOI:10.1016/j.nantod.2012.12.004 |
| [25] |
Z. Wang, G. Liu, H. Zheng, X. Chen, Biotechnol. Adv. 32(2014) 831-843. DOI:10.1016/j.biotechadv.2013.08.020 |
| [26] |
A. Banerjee, T. Pons, N. Lequeux, B. Dubertret, Interface Focus 6(2016) 20160064. DOI:10.1098/rsfs.2016.0064 |
| [27] |
P.E. Boukany, Y. Wu, X. Zhao, et al., Adv. Healthc. Mater. 3(2014) 682-689. DOI:10.1002/adhm.201300213 |
| [28] |
T. Endres, M. Zheng, A. Kılıç, et al., Mol. Pharm. 11(2014) 1273-1281. DOI:10.1021/mp400744a |
| [29] |
J. Park, J. Lee, J. Kwag, et al., ACS Nano 9(2015) 6511-6521. DOI:10.1021/acsnano.5b02357 |
| [30] |
G. Lin, T. Chen, J. Zou, et al., Front. Pharmacol. 8(2017) 182. |
| [31] |
J.M. Li, M.X. Zhao, H. Su, et al., Biomaterials 32(2011) 7978-7987. DOI:10.1016/j.biomaterials.2011.07.011 |
| [32] |
H. Zhu, S. Zhang, Y. Ling, et al., J. Control. Release 220(2015) 529-544. DOI:10.1016/j.jconrel.2015.11.017 |
| [33] |
J. Zhao, X. Qiu, Z. Wang, et al., OncoTargets Ther. 6(2013) 303-309. |
| [34] |
T. Getz, J. Qin, I.L. Medintz, et al., J. Neurochem. 139(2016) 872-885. DOI:10.1111/jnc.2016.139.issue-5 |
| [35] |
Y. Wang, C. Yang, R. Hu, et al., Biomater. Sci. 3(2015) 192-202. DOI:10.1039/C4BM00306C |
| [36] |
Y. Wang, B. Wu, C. Yang, et al., Small 12(2016) 534-546. DOI:10.1002/smll.v12.4 |
| [37] |
G. Chen, J.Y. Zhu, Z.L. Zhang, et al., Angew. Chem. Int. Ed. 54(2015) 1036-1040. DOI:10.1002/anie.201410223 |
| [38] |
M.Z. Zhang, C. Li, B.Y. Fang, et al., Nanotechnology 25(2014) 255102. DOI:10.1088/0957-4484/25/25/255102 |
| [39] |
M.Z. Zhang, Y. Yu, R.N. Yu, et al., Small 9(2013) 4183-4193. DOI:10.1002/smll.v9.24 |
| [40] |
Y. Wu, B. Zhou, F. Xu, et al., Acta Biomater. 46(2016) 165-176. DOI:10.1016/j.actbio.2016.09.008 |
| [41] |
A.M. Derfus, A.A. Chen, D.H. Min, E. Ruoslahti, S.N. Bhatia, Bioconjug. Chem. 18(2007) 1391-1396. DOI:10.1021/bc060367e |
| [42] |
P. Subramaniam, S.J. Lee, S. Shah, et al., Adv. Mater. 24(2012) 4014-4019. DOI:10.1002/adma.v24.29 |
| [43] |
N. Singh, A. Agrawal, A.K. Leung, P.A. Sharp, S.N. Bhatia, J. Am. Chem. Soc. 132(2010) 8241-8243. DOI:10.1021/ja102132e |
| [44] |
J.M. Li, Y.Y. Wang, M.X. Zhao, et al., Biomaterials 33(2012) 2780-2790. DOI:10.1016/j.biomaterials.2011.12.035 |
| [45] |
J. Li, W.Y. Lee, T. Wu, et al., Adv. Healthc. Mater. 5(2016) 1049-1057. DOI:10.1002/adhm.201500879 |
| [46] |
L.E. Rikans, T. Yamano, J. Biochem. Mol. Toxicol. 14(2000) 110-117. DOI:10.1002/(ISSN)1099-0461 |
| [47] |
B.B. Manshian, J. Jimenez, U. Himmelreich, S.J. Soenen, Biomaterials 127(2017) 1-12. DOI:10.1016/j.biomaterials.2017.02.039 |
| [48] |
Z. Hu, B. Song, L. Xu, et al., Biomaterials 108(2016) 187-196. DOI:10.1016/j.biomaterials.2016.08.047 |
| [49] |
T. Zhang, J.L. Stilwell, D. Gerion, et al., Nano Lett. 6(2006) 800-808. DOI:10.1021/nl0603350 |
| [50] |
A.A. Mansur, H.S. Mansur, S.M. de Carvalho, et al., Int. J. Nanomed. 11(2016) 4669-4690. DOI:10.2147/IJN |
| [51] |
K. Peynshaert, S.J. Soenen, B.B. Manshian, et al., Acta Biomater. 48(2017) 195-205. DOI:10.1016/j.actbio.2016.10.022 |
| [52] |
C. Fu, L. Qiang, T. Liu, et al., J. Mater. Chem. B 2(2014) 6978-6983. DOI:10.1039/C4TB01004C |
| [53] |
H. Shi, J. Wei, L. Qiang, X. Chen, X. Meng, J. Biomed. Nanotechnol. 10(2014) 2677-2699. DOI:10.1166/jbn.2014.1881 |
| [54] |
J. Wei, J. Ren, J. Liu, et al., Biosens. Bioelectron. 52(2014) 304-309. DOI:10.1016/j.bios.2013.09.006 |
| [55] |
Q. Wang, C. Zhang, G. Shen, et al., J. Nanobiotechnol. 12(2014) 58. DOI:10.1186/s12951-014-0058-0 |
| [56] |
J. Kim, J. Park, H. Kim, K. Singha, W.J. Kim, Biomaterials 34(2013) 7168-7180. DOI:10.1016/j.biomaterials.2013.05.072 |
| [57] |
L. Hu, Y. Sun, S. Li, et al., Carbon 67(2014) 508-513. DOI:10.1016/j.carbon.2013.10.023 |
| [58] |
L. Wang, X. Wang, A. Bhirde, et al., Adv. Healthc. Mater. 3(2014) 1203-1209. DOI:10.1002/adhm.v3.8 |
| [59] |
S. Das, N. Debnath, Y. Cui, J. Unrine, S.R. Palli, ACS Appl. Mater. Interfaces 7(2015) 19530-19535. DOI:10.1021/acsami.5b05232 |
| [60] |
P. Pierrat, R. Wang, D. Kereselidze, et al., Biomaterials 51(2015) 290-302. DOI:10.1016/j.biomaterials.2015.02.017 |
| [61] |
Y.F. Wu, H.C. Wu, C.H. Kuan, et al., Sci. Rep. 6(2016) 21170. DOI:10.1038/srep21170 |
| [62] |
S. Chen, X. Hao, X. Liang, et al., J. Biomed. Nanotechnol. 12(2016) 1-27. DOI:10.1166/jbn.2016.2122 |
| [63] |
E. Galdiero, A. Falanga, A. Siciliano, et al., Int. J. Nanomed. 12(2017) 2717-2731. DOI:10.2147/IJN |
| [64] |
J. Qiu, R. Zhang, J. Li, et al., Int. J. Nanomed. 10(2015) 6709-6724. |