 2017, Vol. 28
2017, Vol. 28 



b Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
The field of organocatalysis [1] has expanded in recent years, since the discovery of the proline-catalyzed aldol reaction [2]. During this period, an overwhelming number of novel, highly efficient organocatalysts have been reported in the literature. In particular, chiral secondary amines are extremely powerful reagents that have dominated the field of amino catalysis [3]. However, in spite of the tremendous success of the use of secondary amines in the asymmetric functionalization of aldehydes, only minor progress has been achieved in the corresponding transformations of ketones, because of the inherent difficulties in generating congested covalent intermediates between chiral secondary amines and ketones. Primary amine catalysis offers the unique possibility of participating in processes between sterically demanding partners [4] and chiral primary amines have also been demonstrated to be effective catalysts in a wide range of enantioselective organic reactions [5], especially for the activation of challenging substrates, such as α, α-disubstituted aldehydes [6] and ketones [7].
β-Amino carbonyl derivatives bearing heterocyclic nitrogen atoms have become attractive targets in organic and medicinal chemistry, as they are versatile synthetic intermediates that can be used in the preparation of a wide variety of heterocycles [8]. Among the β-amino carbonyl molecules, pyrrolidine and piperidine moieties are extremely valuable scaffolds due to their widespread occurrence in diverse biologically active natural products and pharmaceutical agents [9], as well as their utility as chiral auxiliaries and chiral ligands in asymmetric catalysis [10]. The aza-Michael reaction is the most direct method for selectively creating a carbon–nitrogen bond at the β-position of an activated olefin. Despite the importance of this methodology, organocatalytic enantioselective aza-Michael reactions remained undeveloped until very recently [11] and can thus be considered to be challenging Moreover, most of the present work has focused on intermolecular aza-Michael reactions and only a few reports involving enantioselective organocatalytic intramolecular reactions have been published, especially for α, β-unsaturated ketones.
As part of our continuing interest in the development of a general and highly selective methodology for an intramolecular aza-Michael reaction (IMAMR), herein we described an operationally simple, very high enantioselective organocatalytic IMAMR for carbamates bearing an α, β-unsaturated ketone using chiral primary amines and a weak organic acid as co-catalyst. The application of this methodology for the synthesis of several enantiomeric five-and six-membered heterocycles was also conducted. Starting carbamates, sulfonamides and acetamides bearing conjugated ketones 1 were easily obtained in moderate to high yields through a cross-metathesis reaction of the corresponding unsaturated N-protected amines with vinyl ketones in the presence of second-generation Hoveyda-Grubbs catalyst.
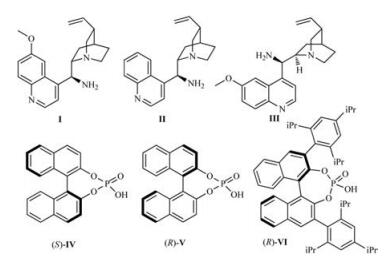
2. Results and discussionIn order to find the optimum conditions and catalysts for the enantioselective IMAMR, the model reaction was performed with catalyst Ⅰ (Fig. 1) and various organic acids as co-catalyst in CHCl3 for 40 h at room temperature (Table 1, entries 1–5). The results indicated that low yields were achieved when weak acids, such as CH3COOH, 2-F-C6H4COOH and 2-NO2-C6H4COOH, were used (Table 1, entries 1–3). A significant improvement in yield (90%) and ee (75%) was achieved when the reaction was performed in the presence of trifluoroacetic acid, and a higher yield (95%) and ee value (89%) of 2a was observed when the slightly weaker acid, DPP (diphenyl hydrogen phosphate), was used (Table 1, entries 4–5). Further screening showed that toluene was a suitable solvent (Table 1, entry 7). Interestingly, in contrast to a previous report [12], the use of a chiral co-catalyst, such as (S)-Ⅳ, (R)-Ⅴ and (R)-Ⅵ (Fig. 1), had a negative effect on both the enantioselectivity and yield of the reactions (Table 1, entries 10–12). Furthermore, increasing the co-catalyst loading from 15 mol% to 30 mol% resulted in a slight decrease in yield (96%) and ee (94%) (Table 1, entry 14). When a lower temperature was used, however, there was no improvement in the final ee (81%) and yield (65%) (Table 1, entry 13). Under the same condition, no improvement in the ee and yield was observed when catalysts Ⅱ and Ⅲ were used (Table 1, entries 15 and 16). Meanwhile, the yield of 2a was decreased when the loading of catalyst Ⅰ was reduced (Table 1, entries 17 and 18).

|
Download:
|
| Fig. 1. Structures of catalysts and organic acids used in this report | |
|
|
Table 1 Catalyst screening and optimization of reaction conditions |
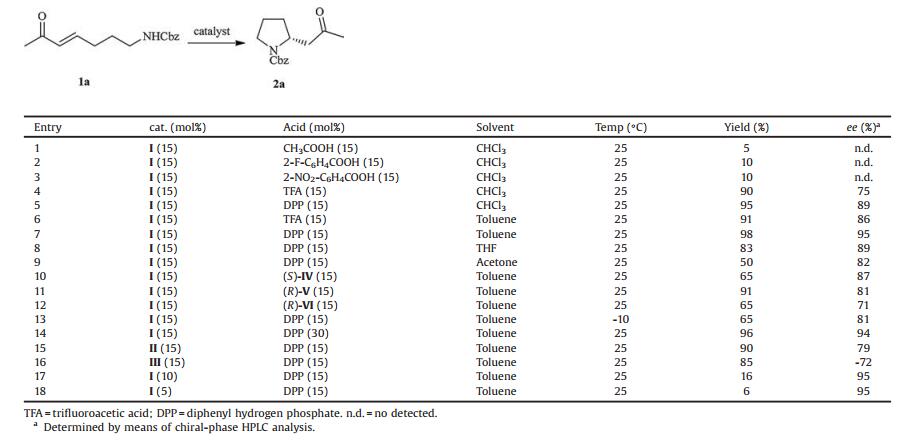
With the optimized conditions in hand, to further explore the influence of protecting groups on the enantioselectivity and yield of the IMAMR (Table 2), substrates 1b–1e, bearing carbamates, sulfonamides, or acetamides as nitrogen-protecting groups, were investigated. Excellent asymmetric induction and yield were observed when Cbz, Boc and Ts were used as protecting groups (Table 2, entries 1–3). Even when substrate 1e, which has very weak nucleophilicity of the acetyl amide [12d], was tested, the IMAMR still provided the desired product 2e with excellent yield (95%) and ee (93%). Unlike previous reports [13], our current methodology suggested that placing a heteroatom in the α-position relative to the nitrogen-centered nucleophile (α-effect) to enhance nucleophilicity was unnecessary. More importantly, this method allowed a highly enantioselective assembly of a stereogenic nitrogen-containing carbon center in the functionalized 2-substituted piperidines, providing an efficient synthetic method for many biologically active 2-substituted piperidine alkaloids [14].
|
|
Table 2 Influence of the nitrogen-protecting group on the IMAMR |
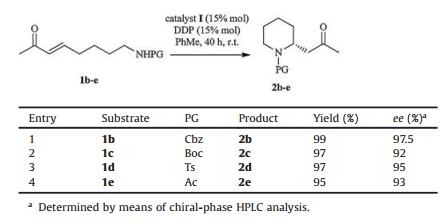
Our attention was then turned to other substrates containing different heteroatoms within the alkyl chain, since the corresponding products could be potentially valuable in medicinal chemistry. Table 3 shows that under the same conditions, reaction of substrates 1f–1i, which contain oxygen, and sulfur in the alkyl chain, led to the corresponding five-or six-membered heterocycles in yields ranging from 55% to 97% and with excellent ee (92%–97%) (Table 3).
|
|
Table 3 Scope of the organocatalytic intramolecular aza-Michael reaction |
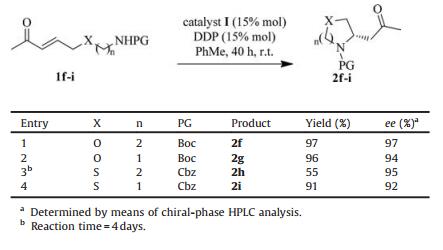
{kind=link}
Finally, the absolute configuration of the newly created stereocenter was determined to be R by comparing the [α]D25 values and NMR data of compound 2a–2c with those described in the literatures [15–17] and identical stereochemical development was assumed for 2d–2i.
3. ConclusionIn conclusion, we have developed a highly efficient and enantioselective organocatalytic IMAMR of carbamates bearing an α, β-unsaturated ketone using 9-amino-9-deoxy-epi-quinine and DPP as co-catalysts. Importantly, a series of synthetically useful 2-substituted five-and six-membered heterocycles could be directly produced in a highly enantioselective fashion (92%–97.5% ee). It is worth noting that in the IMARMR described herein, no α-effect was necessary to enhance the nucleophilicity of the nitrogen and the process took place with very high yield and excellent ee values even if acetamide was used as a nitrogen-protecting group.
4. ExperimentalAll solvents and reagents were used as purchased and received from Aldrich without any further purification. Mass spectra and high-resolution mass spectra were measured on a Finnigan MAT-95 mass spectrometer. 1H NMR and 13C NMR spectra were determined on Bruker AM-400 instrument using tetramethylsilane as an internal reference. Data are presented as follows: chemical shift, multiplicity, coupling constant in hertz (Hz). The signals of the 13C NMR were assigned using DEPT experiments and on the basis of previously published data. The enantiomeric excess (ee) was measured by HPLC (chiralcel OD-H, n-hexane/i-PrOH = 97/ 3, flow rate 1.5 mL/min). Silica gel 60H (200–300 mesh) manufactured by Qingdao Haiyang Chemical Group Co., China) was used for general chromatography.
General procedure for the organocatalytic IMAMR reaction at room temperature: The chiral primary amine Ⅰ (0.015 mmol) was added to a solution of DPP (0.015 mmol) in toluene (0.6 mL) and the mixture was stirred for 15 min at room temperature. Then substrate 1 (0.1 mol) was added. When TLC indicated there was no remaining starting substrate 1, the solvent was removed and the residue was purified by flash column chromatography on silica gel (n-hexane:AcOEt = 7:1), affording the aza-Michael adducts 2a–i.
1H NMR and 13C NMR spectra for all compounds are available in Supporting information and typical spectral data of some compounds are listed below.
Benzyl 2-(2-oxopropyl)pyrrolidine-1-carboxylate (2a): Yield: 98%. [α]D25 +35.5 (c 0.15, CHCl3). ee 95%. 1H NMR (CDCl3, 400 MHz): δ 7.34 (m, 5H), 5.12 (s, 2H), 4.22 (m, 1H), 3.42 (m, 2H), 3.24-2.84 (m, 1H), 2.42 (dd, 1H, J = 16.4, 9.6 Hz), 2.22-2.00 (m, 4H), 1.90–1.78 (m, 2H), 1.70-1.60 (m, 1H); Major: 13C NMR (CDCl3, 100 MHz): δ 207.2, 154.6, 136.8, 128.4, 128.4, 127.9, 127.9, 127.8, 66.5, 53.9, 47.5, 46.3, 30.8, 30.3, 23.6; Minor: 13C NMR (CDCl3, 100 MHz): δ 207.2, 154.6, 137.6, 128.4, 128.4, 127.9, 127.9, 127.8, 66.8, 53.2, 48.5, 46.6, 31.6, 30.3, 22.8; HRMS (EI) calcd. for C15H19NO3 (M+): 261.1365, found 261.1369.
Benzyl 2-(2-oxopropyl)piperidine-1-carboxylate (2b): Yield: 99%. [α]D25 +14.0 (c 0.13, CHCl3). ee 97.5%. 1H NMR (CDCl3, 400 MHz): δ 7.34 (m, 5H), 5.11 (d, 2H, J = 3.2 Hz), 4.80 (m, 1H), 4.03 (m, 1H), 2.85 (t, 1H, J = 12.2 Hz), 2.68 (m, 2H), 2.13 (s, 3H), 1.52 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 206.7, 155.2, 136.7, 128.4, 128.4, 127.9, 127.8, 127.8, 67.0, 47.4, 44.2, 39.7, 30.0, 28.2, 25.1, 18.7; HRMS (EI) calcd. for C16H21NO3 (M+): 275.1521, found 275.1526.
tert-Butyl 2-(2-oxopropyl)piperidine-1-carboxylate (2c): Yield: 97%. [α]D25 +7.3 (c 0.1, CHCl3). ee 92%. 1H NMR (CDCl3, 400 MHz): δ 4.72 (brs, 1H), 3.97 (m, 1H), 2.77 (t, J = 12.4 Hz, 1H), 2.64 (dd, J = 7.1, 2.8 Hz, 2H), 2.18 (s, 3 H), 1.70–1.45 (m, 5H), 1.43 (s, 9H), 1.43–1.40 (m, 1H); 13C NMR (CDCl3, 100 MHz): δ 207.1, 154.7, 79.6, 47.2, 44.3, 39.3, 30.0, 28.3, 28.3, 28.3, 25.2, 18.8; HRMS (EI) calcd. for C13H23NO3 (M+): 241.1678, found 241.1672.
1-(1-Tosylpiperidin-2-yl)propan-2-one (2d): Yield: 97%. [α]D25 +23.0 (c 0.18, CHCl3). ee 95%. 1H NMR (CDCl3, 400 MHz): δ 7.70 (d, 2H, J = 8.2 Hz), 7.27 (d, 2H, J = 8.2 Hz), 4.51 (m, 1H), 3.78 (m, 1H), 2.92 (td, 1H, J = 13.6, 2.4 Hz), 2.78 (dd, 1H, J = 16.0, 9.6 Hz), 2.59 (dd, 1H, J = 16.0, 4.4 Hz), 2.41 (s, 3H), 2.12 (s, 3H), 1.55–1.25 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 206.0, 143.1, 138.0, 129.6, 129.6, 127.0, 127.0, 48.6, 43.8, 41.1, 30.2, 27.6, 24.5, 21.4, 18.3; HRMS (EI) calcd. for C15H21NO3S (M+): 295.1242, found 295.1251.
1-(1-Acetylpiperidin-2-yl)propan-2-one (2e): Yield: 95%. [α]D25 +44.0 (c 0.2, CHCl3). ee 93%. Major: 1H NMR (CDCl3, 400 MHz): δ 5.26 (m, 1H), 3.59 (d, 1H, J = 18.0 Hz), 3.12 (dt, 1H, J = 18.0, 3.2 Hz), 2.79 (d, 1H, J = 9.2 Hz), 2.63 (t, 1H, J = 9.2 Hz), 2.20 (s, 3H), 2.04 (s, 3H), 1.75-1.30 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 207.3, 169.4, 44.7, 44.2, 42.1, 29.8, 28.1, 25.8, 21.9, 18.8; Minor: 1H NMR (CDCl3, 400 MHz): δ 4.62–4.48 (m, 2H), 2.79 (d, 1H, J = 9.2 Hz), 2.63 (t, 1H, J = 9.2 Hz), 2.57–2.47 (m, 1H), 2.17 (s, 3H), 2.14 (s, 3H), 1.75–1.30 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 205.9, 169.4, 49.2, 44.1, 36.9, 30.9, 29.3, 25.3, 21.5, 19.2; HRMS (EI) calcd. for C10H17NO2 (M+): 183.1259, found 183.1266.
tert-Butyl 3-(2-oxopropyl)morpholine-4-carboxylate (2f): Yield: 97%. [α]D25 +38.5 (c 0.15, CHCl3). ee 97%. 1H NMR (CDCl3, 400 MHz): δ 4.38 (brs, 1H), 3.85–3.67 (m, 3H), 3.56 (dd, 1H, J = 12.0, 2.0 Hz), 3.43 (td, 1H, J = 11.7, 2.4 Hz), 3.06 (brs, 2H), 2.56 (d, 1H, J = 15.3 Hz), 2.18 (s, 3H), 1.44 (s, 9H); 13C NMR (CDCl3, 100 MHz):δ 206.5, 154.3, 80.2, 68.9, 66.7, 47.2, 42.4, 39.1, 30.4, 28.2, 28.2, 28.2; HRMS (EI) calcd. for C12H21NO4 (M+): 243.1471, found 243.1472.
tert-Butyl 4-(2-oxopropyl)oxazolidine-3-carboxylate (2g): Yield: 96%. [α]D25 +67.0 (c 0.11, CHCl3). ee 94%. 1H NMR (CDCl3, 400 MHz): δ 4.90–4.65 (m, 2H), 4.20–4.10 (m, 2H), 3.72–3.66 (m, 1H), 3.25–2.85 (m, 1H), 2.65–2.45 (m, 1H), 2.13 (s, 3H), 1.43 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 206.8, 152.5, 80.4, 78.6, 72.5, 51.4, 46.0, 30.2, 28.3, 28.2, 28.3; HRMS (EI) calcd. for C11H19NO4 (M+): 229.1314, found 229.1323.
Benzyl 3-(2-oxopropyl)thiomorpholine-4-carboxylate (2h): Yield: 55%. [α]D25 +18.5 (c 0.1, CHCl3). ee 95%. 1H NMR (CDCl3, 400 MHz): δ 7.35 (m, 5H), 5.13 (s, 2H), 4.99 (brs, 1H), 4.34 (m, 1H), 3.45–2.90 (m, 3H), 2.80–2.30 (m, 4H), 2.16 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 206.1, 155.1, 136.4, 128.5, 128.5, 128.1, 127.9, 127.9, 67.5, 46.4, 43.0, 40.7, 31.1, 30.4, 27.4; HRMS (EI) calcd. for C15H19NO3S (M+): 293.1086, found 293.1089.
(S)-Benzyl 4-(2-oxopropyl)thiazolidine-3-carboxylate (2i): Yield: 91%. [α]D25 +14.2 (c 0.18, CHCl3). ee 92%. 1H NMR (CDCl3, 400 MHz): δ 7.45–7.28 (m, 5H), 5.13 (s, 2H), 4.70–4.45 (m, 2H), 4.33 (d, 1H, J = 9.2 Hz), 3.24 (dd, 1H, J = 12.0, 6.4 Hz), 3.03–2.70 (m, 3H), 2.18-2.02 (m, 3H); Major: 13C NMR (CDCl3, 100 MHz): δ 206.5, 153.4, 136.1, 128.5, 128.5, 128.1, 127.9, 127.9, 67.3, 56.3, 47.8, 45.9, 35.3, 30.2; Minor: 13C NMR (CDCl3, 100 MHz): δ 206.5, 153.4, 136.1, 128.5, 128.5, 128.1, 127.9, 127.9, 67.3, 55.3, 48.5, 46.7, 36.3, 30.2; HRMS (EI) calcd. for C14H17NO3S (M+): 279.0929, found 279.0936.
AcknowledgementWe greatly acknowledge financial support from the National Natural Science Foundation of China (No. 21262022).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at 10.1016/j.cclet.2017.04.017.
| [1] |
(a) P. I. Dalko, L. Moisan, In the golden age of organocatalysis, Angew. Chem. Int. Ed. 43(2004) 5138-5175; (b) A. Berkessel, H. Groger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, Germany, 2005; (c) Lelais, D. W. C. MacMillan, Modern strategies in organic catalysis: the advent and development of iminium activation, Aldri-Chim. Acta 39(2006) 79-87; (d) K. N. Houk, Benjamin List, Asymmetric organocatalysis, Acc. Chem. Res. 37(2004) 487-487. |
| [2] |
(a) B. List, R. A. Lerner, C. F. Barbas Ⅲ, Proline-catalyzed direct asymmetric Aldol reactions, J. Am. Chem. Soc. 122(2000) 2395-2396; (b) U. Eder, G. Sauer, R. Wiechert, New type of asymmetric cyclization to optically active steroid CD partial structures, Angew. Chem. Int. Ed. Engl. 10(1971) 496-497; (c) Z. G. Hajos, D. R. Parrish, Asymmetric synthesis of bicyclic intermediates of natural product chemistry, J. Org. Chem. 39(1974) 1615-1621. |
| [3] |
(a) S. Mukherjee, J. W. Yang, S. Hoffmann, B. List, Asymmetric enamine catalysis, Chem. Rev. 107(2007) 5471-5569; (b) A. Erkkilä, I. Majander, P. M. Pihko, Iminium catalysis, Chem. Rev. 107(2007) 5416-5470. |
| [4] | Xu L.W., Luo J., Lu Y.X.. Asymmetric catalysis with chiral primary amine-based organocatalysts. Chem. Commun. (2009) 1807–1821. |
| [5] |
(a) L. Jiang, Y. C. Chen, Recent advances in asymmetric catalysis with cinchona alkaloid-based primary amines, Catal. Sci. Technol. 1(2011) 354-365; (b) F. Z. Peng, Z. H. Shao, Advances in asymmetric organocatalytic reactions catalyzed by chiral primary amines, J. Mol. Catal. A 285(2008) 1-13; (c) C. Liu, Y. X. Lu, Primary amine/(+)-CSA salt-promoted organocatalytic conjugate addition of nitro esters to enones, Org. Lett. 12(2010) 2278-2281; (d) L. T. Dong, R. J. Lu, Q. S. Du, et al. , Highly enantioselective conjugate addition of 1-bromonitroalkanes to alpha, beta-unsaturated ketones catalyzed by 9-amino-9-deoxyepiquinine, Tetrahedron 65(2009) 4124-4129; (e) J. Zhou, V. Wakchaure, P. Kraft, B. List, Primary-amine-catalyzed enantioselective intramolecular aldolizations, Angew. Chem. Int. Ed. 47(2008) 7656-7658; (f) F. Xue, S. L. Zhang, W. H. Duan, W. Wang, A novel bifunctional sulfonamide primary amine-catalyzed enantioselective conjugate addition of ketones to nitroolefins, Adv. Synth. Catal. 350(2008) 2194-2198; (g) S. Z. Luo, H. Xu, L. Zhang, J. Y. Li, J. P. Cheng, Highly enantioselective direct syn-and anti-aldol reactions of dihydroxyacetones catalyzed by chiral primary amine catalysts, Org. Lett. 10(2008) 653-656. |
| [6] |
(a) M. P. Lalonde, Y. Chen, E. N. Jacobsen, A chiral primary amine thiourea catalyst for the highly enantioselective direct conjugate addition of a, a-disubstituted aldehydes to nitroalkenes, Angew. Chem. Int. Ed. 45(2006) 6366-6370; (b) S. H. McCooey, S. J. Connon, Readily accessible 9-epi-amino cinchona alkaloid derivatives promote efficient, highly enantioselective additions of aldehydes and ketones to nitroolefins, Org. Lett. 9(2007) 599-602. |
| [7] |
(a) G. Bartoli, P. Melchiorre, A novel organocatalytic tool for the iminium activation of α, β-unsaturated ketones, Synlett 13(2008) 1759-1772; (b) Y. C. Chen, The development of asymmetric primary amine catalysts based on cinchona alkaloids, Synlett 13(2008) 1919-1930. |
| [8] |
(a) D. O'Hagan, Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids, Nat. Prod. Rep. 17(2000) 435-446; (b) S. Laschat, T. Dickner, Stereoselective synthesis of piperidines, Synthesis (2000) 1781-1813. |
| [9] |
(a) E. C. Juaristi, V. A. Soloshonok, Enantioselective Synthesis of β-Amino Acids, 2nd. ed. , Wiley-VCH Ltd. , New York, 2005; (b) J. W. Daly, T. F. Spande, H. M. Garraffo, Alkaloids from amphibian skin: a tabulation of over eight-hundred compounds, J. Nat. Prod. 68(2005) 1556-1575; (c) M. G. P. Buffat, Synthesis of piperidines, Tetrahedron 60(2004) 1701-1729; (d) P. M. Weintraub, J. S. Sabor, J. M. Kane, D. R. Borcherding, Recent advances in the synthesis of piperidones and piperidines, Tetrahedron 59(2003) 2953-2989; (e) M. D. Groaning, A. I. Meyers, Chiral non-racemic bicyclic lactams. Auxiliarybased asymmetric reactions, Tetrahedron 56(2000) 9843-9873; (f) S. Leclercq, J. C. Braekman, D. Daloze, J. M. Pasteels, The defensive chemistry of ants, Prog. Chem. Org. Nat. Prod. 79(2000) 115-229; (g) J. W. Daly, H. M. Garraffo, T. F. Spende, Alkaloids: chemical and biological perspectives, 14th. ed. , Pergamon, New York, 1999; (h) C. Escolano, M. Amat, J. Bosch, Chiral oxazolopiperidone lactams: versatile intermediates for the enantioselective synthesis of piperidine-containing natural products, Chem. Eur. J. 12(2006) 8198-8207; (i) M. Schneider, Alkaloids: chemical and biological perspectives, 10th. ed. , Pergamon, New York, 1996; (j) H. Xu, H. Tang, H. Feng, Y. Li, Design, synthesis and anticancer activity evaluation of novel C14 heterocycle substituted epi-triptolide, Eur. J. Med. Chem. 73(2014) 46-55. |
| [10] |
(a) B. H. Kim, H. B. Lee, J. K. Hwang, Y. G. Kim, Asymmetric induction in the conjugate addition of thioacetic acid to methacrylamides with chiral auxiliaries, Tetrahedron: Asymmetry 16(2005) 1215-1220; (b) M. Nyerges, D. Bendell, A. Arany, et al. , Silver acetate catalysed asymmetric 13-dipolar cycloadditions of imines and chiral acrylamides, Synlett (2003) 947-950; (c) F. Fache, E. Schultz, M. L. Tomasino, M. Lemaire, Nitrogen-containing ligands for asymmetric homogeneous and heterogeneous catalysis, Chem. Rev. 100(2000) 2159-2232; (d) J. A. Sweet, J. M. Cavallari, W. A. Price, J. W. Ziller, D. V. McGrath, Synthesis and characterization of new amine-imine ligands based on trans-25-disubstituted pyrrolidines, Tetrahedron: Asymmetry 8(1997) 207-211; (e) J. K. Whitesell, C2 symmetry and asymmetric induction, Chem. Rev. 89(1989) 1581-1590. |
| [11] |
(a) D. Enders, C. Wang, J. X. Liebich, Organocatalytic asymmetric aza-Michael additions, Chem. Eur. J. 15(2009) 11058-11076; (b) J. L. Vicario, D. Badia, L. Carrillo, (+)-(S, S)-Pseudoephedrine as a chiral auxiliary in asymmetric Mannich reactions: scope and limitations, Synthesis (2006) 4065-4074; (c) J. L. Vicario, L. Carrillo, J. Etxebarria, E. Reyes, N. Ruiz, The asymmetric azaMichael reaction. a review, Org. Prep. Proced. Int. 37(2005) 513-538; (d) L. W. Xu, C. G. Xia, A catalytic enantioselective aza-Michael reaction: novel protocols for asymmetric synthesis of b-amino carbonyl compounds, Eur. J. Org. Chem. (2005) 633-639; (e) S. Fustero, J. Moscardo, D. Jimenez, et al. , Organocatalytic approach to benzofused nitrogen-containing heterocycles: enantioselective total synthesis of (+)-angustureine, Chem. Eur. J. 14(2008) 9868-9872; (f) M. Bandini, A. Eichholzer, M. Tragni, A. Umani-Ronchi, Enantioselective phase-transfer-catalyzed intramolecular aza-Michael reaction: effective route to pyrazino-indole compounds, Angew. Chem. Int. Ed. 47(2008) 3238-3241; (g) S. Fustero, D. Jimenez, J. Moscardo, S. Catalan, C. del Pozo, Enantioselective organocatalytic intramolecular aza-Michael reaction: a concise synthesis of (+)-sedamine. , (+)-allosedamine, and (+)-coniine, Org. Lett. 9(2007) 5283-5286; (h) K. Takasu, S. Maiti, M. Ihara, Asymmetric intramolecular aza-Michael reaction using environmentally friendly organocatalysis, Heterocycles 59(2003) 51-55; (i) J. D. Liu, Y. C. Chen, G. B. Zhang, et al. , Asymmetric organocatalytic intramolecular aza-Michael addition of enone carbamates: catalytic enantioselective access to functionalized 2-substituted piperidines, Adv. Syn. Catal. 353(2011) 2721-2730; (j) L. Y. Wu, G. Bencivenni, M. Mancinelli, et al. , Organocascade reactions of enones catalyzed by a chiral primary amine, Angew. Chem. Int. Ed. 48(2009) 7196-7199; (k) B. S. Li, E. Zhang, Q. W. Zhang, et al. , One-pot construction of multisubstituted spiro-cycloalkanediones by an organocatalytic asymmetric epoxidation/semipinacol rearrangement, Chem. Asian J. 6(2011) 2269-2272; (l) M. W. Paixao, N. Holub, C. Vila, et al. , Trends in organocatalytic conjugate addition to enones: an efficient approach to optically active alkynyl, alkenyl, and ketone products, Angew. Chem. Int. Ed. 48(2009) 7338-7342; (m) W. Chen, W. Du, Y. Z. Duan, et al. , Enantioselective 13-dipolarcycloaddition of cyclic enones catalyzed by multifunctional primary amines: beneficial effects of hydrogen bonding, Angew. Chem. Int. Ed. 46(2007) 7667-7670. |
| [12] |
(a) O. Lifchits, C. M. Reisinger, B. List, Catalytic asymmetric epoxidation of a-branched enals, J. Am. Chem. Soc. 132(2010) 10227-10229; (b) C. M. Reisinger, X. Wang, B. List, Catalytic asymmetric hydroperoxidation of α, β-unsaturated ketones: an approach to enantiopure peroxyhemiketals, epoxides, and aldols, Angew. Chem. Int. Ed. 47(2008) 8112-8115; (c) X. Tian, C. Cassani, Y. Liu, et al. , Diastereodivergent asymmetric sulfaMichael additions of a-branched enones using a single chiral organic catalyst, J. Am. Chem. Soc. 133(2011) 17934-17941; (d) S. Fustero, S. Monteagudo, M. Sanchez-Rosello, et al. , N-Sulfinyl amines as a nitrogen source in the asymmetric intramolecular aza-Michael reaction: total synthesis of (-)-pinidinol, Chem. Eur. J. 16(2010) 9835-9845. |
| [13] | Heaton M.M.. Quantum mechanical studies of the alpha effect. J. Am. Chem. Soc. 100 (1978) 2004–2008. DOI:10.1021/ja00475a005 |
| [14] |
(a) E. C. Carlson, L. K. Rathbone, H. Yang, N. D. Collett, R. G. Carter, Improved protocol for asymmetric, intramolecular heteroatom Michael addition using organocatalysis: enantioselective syntheses of homoproline, pelletierine, and homopipecolic acid, J. Org. Chem. 73(2008) 5155-5158; (b) P. Macours, J. C. Braekman, D. Daloze, Concise asymmetric syntheses of (+)-tetraponerine-8 and (-)-tetraponerine-8, (+)-tetraponerine-7 and (-)-tetraponerine-7, and theirethyl homologs. a correctionof the structuresof tetraponerine-3, and tetraponerine-7, Tetrahedron 51(1995) 1415-1428. |
| [15] | Majik M.S., Tilve S.G.. Syntheses of (-)-hygrine and (-)-norhygrine via wacker oxidation. Tetrahedron Lett. 51 (2010) 2900–2902. DOI:10.1016/j.tetlet.2010.03.098 |
| [16] | Takahata H., Kubota M., Takahashi S., Momose T.. A newasymmetric entry to 2-substituted piperidines. A concise synthesis of (+)-coniine, (-)-pelletierine, (+)-delta-coniceine, and (+)-epidihydropinidine. Tetrahedron:Asymmetry 7 (1996) 3047–3054. DOI:10.1016/0957-4166(96)00395-3 |
| [17] |
(a) S. G. Davies, A. M. Fletcher, P. M. Roberts, A. D. Smith, Asymmetric synthesisof sedum alkaloids via lithium amide conjugate addition, Tetrahedron 65(2009) 10192-10213; (b) I. Coldham, D. Leonori, Regioselective and stereoselective copper(I)-promoted allylation and conjugate addition of N-Boc-2-lithiopyrrolidine and N-Boc-2-lithiopiperidine, J. Org. Chem. 75(2010) 4069-4077. |