 2017, Vol. 28
2017, Vol. 28 



b State Key Laboratory of Oral Disease, West China School of Stomatology, Sichuan University, Chengdu, 610041, China
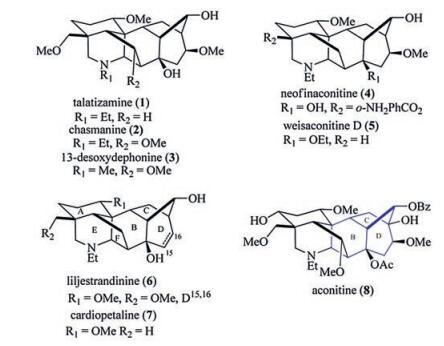
C19-Diterpenoid alkaloids are a large family of highly complex polycyclic alkaloids, which are characterized by an intricate hexacyclic ring system, heavily substituted by a series of oxygen functions such as hydroxy, methoxy and acyloxy groups (Fig. 1) [1]. More than 600 family members have been isolated over the past 60 years. A survey of an extensive number of these alkaloids reveals anti-arrhythmic, anti-inflammatory, anti-epileptic, hypotensive, and bradycardic properties. This broad spectrum of biological activity can be attributed to the potent interactions of these alkaloids with voltage-dependent sodium, potassium, and calcium ion channels [2]. While the toxicity of the most of active compounds has so far limited their clinical application, less toxic derivatives are emerging that could prove to be promising drug candidates [3].

|
Download:
|
| Fig. 1. Structures of C19-diterpenoid alkaloids | |
{kind=link}
The structural intricacy and biological significance of C19-diterpenoid alkaloids have attracted the great attention of many synthetic chemists over the last fifty decades. Early important and impressive contributions to the syntheses of the C19-diterpenoid alkaloids including a synthesis of talatizamine 1, chasmanine 2, and 13-desoxydephonine 3 were made by Wiesner and coworkers in 1970's [4]. Recently, an elegant synthesis of neofinaconitine 4 was completed by the group of Gin using a highly convergent strategy [5]. Subsequently, Sarpong's group and Fukayama's group reported the total syntheses of liljestrandinine 6 and weisaconitine D 5 [6] and the first asymmetric total synthesis of (-)-cardiopetaline 7 [7] respectively, both of which employ a late-staged Wagner-Meerwein rearrangement from a C20-DA skeleton as the key transformation. Despite numerous other synthetic efforts [8, 9], these compounds 1-7 remain the only seven natural C19-diterpenoid alkaloid molecules completed by total syntheses to date. Some C19-diterpenoid alkaloids with more complex oxygen functions and distinguished biological activities, e.g., aconitine 8 and methyllycaconitine, are still the highly attractive and challenging targets for total synthesis.
In view of our long-standing interest toward the chemistry of C19-diterpenoid alkaloids with significant biological activities [9], we planned to initiate a new program to study the total synthesis of aconitine 8. To achieve this goal, we conceived that it was necessary to develop an efficient and scalable synthesis for the rapid construction of the unique tricyclic subunit of tricyclo [6.2.1.0] undecane core 12. Our whole synthetic plan based on this tricyclic skeleton is briefly outlined in Scheme 1, We envisioned that aconitine 8 can be firstly transformed to intermeidate 9 by means of a serials of functional groups interconversions. Then disconnections of C4-C5 and C3-C4 bond involving sequential 6-exo-trig radical cyclization [10] and Michael addition could greatly simplify the hexacyclic molecule into the less complicated tetracyclic structure 10, which might be further degraded into structure 11. With a successive alkylation and intramolecular aldol reaction strategy [11], 11 could be generated from 12 byinstallation of the five-membered ring F on ring B. Thus, development of a highly efficient synthetic route to access this tricyclic intermediate 12 is fairly crucial to realize our whole synthetic strategy.

|
Download:
|
| Scheme1. Retrosynthetic analysis of aconitine | |
{kind=link}
Herein, we wish to report a short sequence to prepare this key tricyclic skeleton using an unprecedented oxidative dearomatization/dimerization/retro-DA/IMDA cascade reaction and WagnerMeerwein rearrangement as the key steps.
2. Results and discussionAs illustrated in Scheme 2, we began our synthetic study with the preparation of the phenol 21 tethering an oxygen substituted olefinic side chain. Thus, coupling 17 with the iodide derivative in presence of n-butyl lithium provided the 18. Removal of TBS protecting group followed by oxidation of primary alcohol with Dess-Martin periodinane afforded the aldehyde 19 in 90% yield. Barbier's allylation [12] of aldehyde 19 with allyl bromide in the presence of Zn/NH4Cl followed by removal of the THP group under acidic condition furnished the allylic alcohol 20. Finally, selective protection of allylic alcohol with TBS provided the phenol 21, ready for the next crucial oxidative dearomatization/intramolecular Diels-Alder (IMDA) cascade reaction. Initial execution of phenol 21 to the oxidative condition by treatment with diacetoxyiodobenzene (PIDA) in a diluted methanonic solution at reflux gave the desired cycloadduct 23 as minor product while the intermolecular Diels-Alder cycloadduct dimer 22 as the major product [9b]. Gratifyingly, we next found this dimer 22 could be successfully converted to the cycloadduct 23 in excellent yield through a retroDA/IMDA process under a thermodynamic condition (heating at 180 ℃ in mesitylene) [13]. Thus, a cascade process involving oxidative dearomatization/dimerization/retro-DA/IMDA reaction could be actually applied to this transformation in one pot. Upon complete formation of dimer 22 by exposure of 21 to PIDA in methanol at low temperature, simply switch the solvent to mesitylene followed by heating at 180 ℃ directly led to the desired cycloadduct 22 in excellent yield and fairly good diastereoselectivity. The stereochemistry of 22 was assigned on the basis of the established endo cycloaddition mechanism [13a] as well as the observed NOE correlation between the methyl of TBS group and the olefin proton. It is worthwhile to mention that the high diastereoselectivity of this reaction make it possible to realize enantioselective synthesis of this important intermediate, as long as construction of the asymmetric chiral center of the secondary alcohol 20.

|
Download:
|
| Scheme2. Synthesis of BCD ring system 26. Reagents and conditions: (a) n-BuLi, THF, 0 ℃–r.t., 2h, 82%; (b) TBAF, THF, r.t., 30min, 97%; (c) DMP, NaHCO3, DCM, r.t., 30min, 90%; (d) allyl bromide, zinc powder, THF/sat. aq NH4Cl (1:4v/v), r.t., 30min; (e) 3mol/L aqueous HCl, MeOH, r.t., 1h, 87% over two steps; (f) TBSOTf, 2, 6-lutidine, DCM, 0 ℃, 1h, 80%; (g) PIDA, MeOH, 0 ℃–r.t., 4h, then mesitylene, 180 ℃, 4h, 90%; (h) LiAlH4, THF, 0 ℃, 3h, 40% for 24b and 50% for 24a; (i) oxalyl chloride, DMSO, Et3N, DCM, -78 ℃ -30 ℃, 1h, 94%; (j) Tf2O, pyridine, DCM, 0 ℃, 1h then silica gel, 85% | |
{kind=link}
With the requisite tricycle functionality in place, the deformation of the [2.2.2]octane subunit of 23 to the [3.2.1] subunit by means of the Wagner-Meerwein rearrangment was addressed next. To realize this desired transformation, it is requisite to reduce the carbonyl group to hydroxy group then sulfonylation to achieve a suitable leaving group with desired α configuration, due to the concerted nature of this kind of rearrangement [9b]. Thus, after preliminary reductive conditions screening involving NaBH4, LiAlH4, Red-Al, LiEt3BH, and Li/NH3, we found the LiAlH4 could give relatively the best result, leading to a separable mixture of stereoisomers 24a and 24b from which the desired α-OH isomer 24a could be isolated in 50% yield. While the isomeric ratio is quite low, the undesired isomer 24b (40% yield) could be separated and cleanly recycled to ketone 23 by Swern oxidation, to maximally avoid the loss of material. With the pure a-OH epimer in place, the Wagner-Meerwein rearrangement was focused next. To our delight, exposure of 24a to Tf2O/pyridine in dichlomethane generated the crude triflate 25, which subsequently underwent a facile rearrangement upon purification by silica gel chromatography, affording the desired tricyclic compound 26 in 85% yield.
3. ConclusionIn summary, we have developed an efficient and concise synthetic approach for the first rapid construction of the unique BCD ring system 26 commonly found in the C19-diterpenoid alkaloids in 21% yield for total nine steps. Keysteps include a highly efficient diastereoselective oxidative dearomatization/dimerization/retro-DA/IMDA cascade reaction and a quite convenient Wagner-Meerwein rearrangement. The double bond and oxygen substituents introduced in ring D of 26 provide the synthetic potential to realize the substitution pattern of aconitine-type diterpenoid alkaloid whereas the oxygen function (OTBS) on ring B woulddeliverampleopportunityfor installationof five-membered ring F and the following ring AE. Further studies toward the total synthesis of aconitine and other members of the C19-diterpenoid alkaloids are currently underway in our laboratory.
4. ExperimentalUnless otherwise indicated, all reactions were carried out under an atmosphere of argon or nitrogen. Unless otherwise noted, all chemicals and solvents were obtained from commercial vendors and used without further purification unless otherwise noted. Anhydrous THF were dried by distillation over sodium and benzophenone. DCM was dried by distillation over CaH2. TLC inspections were carried on silica gel GF254 plates. Column chromatography was performed on silica gel (200–300 mesh, Qingdao Marine Chemical Factory, China). 1H NMR and 13C NMR spectra were acquired on a Varian INOVA-400/54 spec-trometer and Agilent Technologies 600/54 Premium with TMS as internal standard, spectra recorded in CDCl3 were referenced to residual CHCl3 at 7.26 ppm for 1H NMRor 77.0ppmfor 13C NMR. HRMS were obtained with a Waters Quattro Premier XE (Waters) mass spectrometer.
4.1. Preparation of compound 18To a solution of 2-methoxyphenol THP ether 17 (2.00g, 9.61mmol) in 48mL THF at 0 ℃ was added n-butyl lithium (7.80mL, 1.6mol/L in hexane, 12.48mmol) dropwised. The resulting mixture was stirred at room temperature for 30min, and then the iodide derivative ICH2CH2OTBS (3.30g, 11.54mmol) was added at 0 ℃. The reaction mixture was warmed to room temperature and stirred for 2h, and then the mixture was quenched with sat. aq NaHCO3 (15mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (4 ×30mL). The combined organic phases were washed with brine (20mL), dried (Na2SO4), concentrated and purified by flash chromatography eluting with petroleum ether/ethyl acetate (15:1) to give compound 18 (light yellowoil, 2.88g, 82%). 1H NMR (400MHz, CD3OD): δ 7.00–6.96 (m, 1H), 6.91–6.85 (m, 1H), 6.81 (d, 1H, J=7.6Hz), 5.16 (m, 1H), 4.09 (m, 1H), 3.91–3.84 (m, 2H), 3.82 (s, 3H), 3.56–3.50 (m, 1H), 3.11–3.03 (m, 1H), 2.86–2.78 (m, 1H), 1.99–1.90 (m, 3H), 1.66–1.59 (m, 3H), 0.91 (s, 9H), 0.03 (d, 6H, J=4.8Hz); 13C NMR (151MHz, CD3OD): δ 153.61, 146.22, 134.06, 124.81, 124.08, 111.92, 102.97, 64.77, 64.63, 56.23, 35.31, 31.98, 26.51, 26.41, 20.99, 19.16, -5.08, -5.09; HRMS (ESI+): m/z calcd. for C20H34O4Si+ 367.2305 [M+H]+, found 367.2300.
4.2. Preparation of aldehyde 19To a stirred solution of 18 (1.18g, 3.22 mmol) in THF (16 mL), TBAF (2.04g, 6.45 mmol) was added and stirred for 30min. After complete conversion as showed by TLC, the reactionwas quenched with sat. aq NaHCO3 (5 mL) and extracted with EtOAc (3 ×20 mL). The combined organic phases was washed with brine, dried with Na2SO4 and concentrated invacuo. The crude materialwas purified by flash chromatography using PE: EtOAc (9:1 →3:1) to give alcohol 19s (yellow oil, 788mg, 97%).
A solution of alcohol 19s (560 mg, 2.22 mmol) in DCM (22 mL) was treated with NaHCO3 (747 mg, 8.89 mmol) and DMP (1.89 g, 4.45 mmol) in turn at room temperature. The reaction mixture was stirred for 30 min and quenched with sat.aq Na2S2O3 (10mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 ×20 mL). The combined organic layers were washed with brine, dried (Na2SO4), concentrated in vacuo and purified by flash chromatography on silica gel eluting with petroleum ether/ ethyl acetate (6:1) to give aldehyde 19 (colorless oil, 500mg, 90%). 1H NMR (400MHz, CDCl3): δ 9.69 (s, 1H), 7.06 (t, 1H, J=8.0Hz), 6.88 (d, 1H, J=8.4Hz), 6.77 (d, 1H, J=7.6Hz), 5.12 (d, 1H, J=4.0Hz), 3.95 (dd, 1H, J=10.8, 5.2Hz), 3.85 (s, 3H), 3.81–3.69 (m, 2H), 3.49 (dd, 1H, J=10.4, 5.2Hz), 2.00–1.79 (m, 3H), 1.66–1.51 (m, 3H); 13C NMR (151 MHz, CDCl3): δ 200.63, 152.45, 144.90, 127.45, 124.40, 122.78, 111.65, 102.07, 64.17, 55.70, 45.39, 30.76, 25.03, 20.10; HRMS (ESI+): m/z calcd. for C14H18O4+ 251.1283 [M+H]+, found 251.1281.
4.3. Preparation of phenol 20A solution of 19 (200mg, 0.80mmol) in THF (2mL) and sat. aq NH4Cl (8mL) was treated with zinc powder (210mg) and allyl bromide (0.28mL, 3.2mmol) in turn at room temperature. The reaction mixture was stirred for 30min at room temperature and quenched with sat.aq NaHCO3 (10mL) at 0 ℃. The layers were separated and the aqueous layer was extracted with DCM (3 ×20mL). The combined organic layers were washed with brine, dried (Na2SO4), concentrated in vacuo and purified by flash chromatography on silica gel eluting with petroleum ether/ethyl acetate (6:1) to give alcohol 20s (yellow oil, 215mg, 92%).
To a stirred solution of 20s (230 mg, 0.79 mmol) in MeOH (7.9 mL), 3 mol/L HCl (0.8 mL, 2.37 mmol) was added at room temperature and stirred for 1h. The reaction was then quenched with sat. aq NaHCO3 (3 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 ×15 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (3:1) to give phenol 20 (yellow oil, 155mg, 95%). 1H NMR (400MHz, CDCl3): δ 6.83–6.71 (m, 3H), 5.93–5.82 (m, 1H), 5.18–5.12 (m, 2H), 4.03–3.95 (m, 1H), 3.88 (s, 3H), 2.91 (dd, 1H, J=14.0, 4.0Hz), 2.78 (dd, 1H, J=14.0, 7.6Hz), 2.39–2.30 (m, 1H), 2.28–2.19 (m, 1H). 13C NMR (151MHz, CDCl3): δ 147.02, 144.02, 134.77, 124.67, 123.45, 119.56, 117.92, 109.40, 71.34, 55.89, 41.30, 37.61. HRMS (ESI+): m/z calcd. for C12H16O3+ 209.1178 [M+H]+, found 209.1175.
4.4. Preparation of silyl ether 212, 6-Lutidine (0.17 mL, 1.44 mmol) and TBSOTf (0.16 mL, 0.72 mmol) was added dropwise to a solution of 20 (100 mg, 0.48 mmol) in DCM (4 mL) at 0 ℃ in turn. The reaction mixture was stirred for 1 h at this temperature, and then the mixture was diluted with sat. aq NH4Cl (6 mL). The organic layers were separated and the aqueous layer was extracted with DCM (3 ×15 mL). The combined organic phases was washed with brine, dried (Na2SO4), concentrated in vacuo and purified by flash chromatography on silica gel eluting with petroleum ether/ethyl acetate (6:1) to give compound 21 (colorless oil, 124 mg, 80%). 1H NMR (400 MHz, CDCl3): δ 6.75 (d, 2H, J = 4.8 Hz), 6.72–6.67 (m, 1H), 6.57 (s, 1H), 5.85 (dt, 1H, J = 17.7, 7.2 Hz), 5.09–5.02 (m, 2H), 4.09– 4.02 (m, 1H), 3.87 (s, 3H), 2.86 (dd, 1H, J = 13.6, 4.8 Hz), 2.77 (dd, 1H, J = 13.6, 6.8 Hz), 2.22 (m, 2H), 0.87 (s, 9H), 0.01 (s, 3H), -0.10 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 147.05, 144.31, 135.01, 124.93, 124.18, 119.07, 117.08, 109.27, 72.23, 55.95, 41.36, 38.37, 25.87, 18.10, -4.82, -5.04; HRMS (ESI+): m/z calcd. for C18H30O3Si+ 323.2042 [M+H]+, found 323.2045.
4.5. Preparation of cycloadduct 23A solution of 21 (50 mg, 0.15 mmol) in MeOH (1.5 mL) was treated with PIDA (64 mg, 0.20 mmol) in one portion at 0 ℃. The resulting mixture was allowed to stir for 4 h at room temperature and the solvent was evaporated under reduced pressure. The residue was dissolved with mesitylene (2 mL) and stirred 4 h at 180 ℃. The solvent was evaporated under reduced pressure by oil pump and the residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (30:1) to give ketone 23 (yellow solid, 49 mg, 90%). 1H NMR (600 MHz, CDCl3): δ 6.43 (t, 1 H, J = 7.4 Hz), 5.90 (d, 1 H, J = 7.8 Hz), 4.33 (m, 1 H), 3.33 (s, 3 H), 3.32 (s, 3 H) 3.06 (m, 1 H), 2.53 (dd, 1 H, J = 13.8, 8.4 Hz), 2.23–2.18 (m, 1 H), 2.16–2.11 (m, 1 H), 2.10–2.03 (m, 1 H), 1.61 (dd, 1 H, J = 13.8, 3.0 Hz), 1.23–1.18 (m, 1 H), 1.12 (ddd, 1 H, J = 12.0, 6.6, 1.8 Hz), 0.87 (s, 9 H), 0.03 (d, 6 H, J = 10.2 Hz); 13C NMR (151 MHz, CDCl3): δ 200.86, 135.19, 133.95, 131.96, 130.89, 94.68, 73.06, 72.75, 59.30, 50.15, 49.87, 42.64, 39.84, 39.71, 38.65, 38.37, 37.08, 26.97, 25.85, 18.02, -4.87; HRMS (ESI+): m/z calcd. for C19H32O4Si+ 353.2148 [M+H]+, found 353.2150.
4.6. Preparation of alcohol 24a and 24bTo a solution of compound 23 (80 mg, 0.23 mmol) in THF (2.4 mL) at 0 ℃ was added LiAlH4 (0.25 ml, 1 mol/L in THF, 0.25 mmol) dropwise. After stirred for 3 h, the reaction was quenched by sat. aq NaOH (10 mL), filtered, and extracted with EtOAc (4 ×15 mL). The combined extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (20:1) to give the alcohol 24a (light yellow oil, 40 mg, 50%) and 24b (light yellow oil, 32 mg, 40%). For 24a: 1H NMR (400 MHz, CDCl3): δ 6.30–6.21 (m, 1 H), 6.00 (d, 1H, J = 8.0 Hz), 4.39– 4.32 (m, 1 H), 3.34 (d, 1H, J = 4.9 Hz), 3.28 (s, 3H), 3.20 (s, 3 H), 2.83– 2.79 (m, 1 H), 2.35–2.24 (m, 2H), 2.12 (m, 2 H), 1.68–1.59 (m, 1 H), 1.26–1.20 (m, 1 H), 1.02 (m, 1 H), 0.86 (s, 9H), 0.01 (d, 6 H, J = 4.8 Hz); 13C NMR (151 MHz, CDCl3): δ 136.08, 130.51, 104.25, 79.86, 73.38, 51.86, 49.70, 49.43, 43.65, 43.18, 39.02, 38.44, 27.90, 25.81, 17.98, -4.85; HRMS (ESI+): m/z calcd. for C19H34O4Si+ 355.2305 [M + H]+, found 355.2302.
4.7. Conversion of 24b to ketone 23To a solution of oxalyl chloride (140 uL, 2 mol/L in DCM, 0.28 mmol) at -78 ℃ was added DMSO (0.5 mL, 2 mol/L in DCM, 1.00 mmol) dropwise, and the resulting mixture was stirred at -78 ℃ for 30 min, and then a solution of 24b (32 mg, 0.09 mmol) in DCM (1 mL) was added dropwise. After 1 h, Et3N (0.2 mL, 1.35 mmol) was added dropwise, and stirred for 2 h, at which point it was warmed to -30 ℃. After 30 min, the reaction mixture was quenched with sat. aq NH4Cl (7 mL) and extracted with DCM (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4 and concentrated to oil. Purification by flash chromatography (PE: EA = 30: 1) provided compound 23 (yellow solid, 30 mg, 94%).
4.8. Preparation of BCD tricyclic ring system 26To a solution of alcohol 24a (36 mg, 0.10 mmol) in DCM (1 mL) was added pyridine (41 mL, 0.51 mmol), Tf2O (34 mL, 0.20 mmol) in turn at 0 ℃. The reaction mixture was stirred for 1 h, and quenched by saturated water (1 mL). The layers were seperated and the aqeous layer was extracted by DCM (4 × 5 mL). The combined organic solution was dried over anhydrous Na2SO4, then filtered and evaporated in vacuo. The residue was purified by flash silical gel column chromatography (Petroleum ether/ethyl acetate, 15:1) to get 26 (yellow oil, 31 mg, 85%). 1H NMR (400 MHz, CDCl3): δ 5.79–5.72 (m, 1 H), 5.51 (d, 1 H, J = 9.6 Hz), 4.15–4.10 (m, 1 H), 3.22 (s, 3 H), 3.19 (s, 3 H), 2.36 (t, 2 H, J = 6.4 Hz), 2.15 (d, 1 H, J = 6.0 Hz), 1.98 (m, 2 H), 1.72 (m, 1 H), 1.67 (s, 2 H), 1.61 (dd, 1 H, J = 13.6, 2.8 Hz), 0.84 (s, 9 H), 0.08 (s, 6 H); 13C NMR (151 MHz, CDCl3): δ 135.32, 126.79, 111.45, 71.94, 67.91, 50.11, 47.68, 47.49, 43.61, 39.90, 35.07, 34.16, 30.35, 25.76, 25.73, 17.94, -4.98, -5.18; HRMS (ESI+): m/z calcd. for C19H34O4Si+ 355.2305 [M + H]+, found 355.2300.
AcknowledgmentsFinancial support for this work was provided by the National Natural Science Foundation of China (No. 21472129 and No. 21272163).
| [1] | F. P. Wang, Q. H. Chen, in: G. A. Cordell (Ed. ), In the Alkaloids: Chemistry and Biology, 69, Elsevier Science, Amsterdam, 2010, pp. 1-577. |
| [2] | A. Ameri. The effects of aconitum alkaloids on the central nervous system. Prog. Neurobiol. 56 (1998) 211–235. DOI:10.1016/S0301-0082(98)00037-9 |
| [3] | K.J. Goodall, D. Barker, M.A. Brimble. A review of advances in the synthesis of analogues of the delphinium-alkaloid methyllycaconitine. Synlett (2005) 1809–1827. |
| [4] |
(a) K. Wiesner, T. Y. R. Tsai, K. Huber, et al. , Total synthesis of talatisamine, a delphinine type alkaloid, J. Am. Chem. Soc. 96(1974) 4990-4992; (b) K. Wiesner, T. Y. R. Tsai, K. P. Nambiar, A new stereospecific total synthesis of chasmanine and 13-desoxydelphonine, Can. J. Chem. 56(1978) 1451-1454; (c) K. Wiesner, Total synthesis of delphinine-type alkaloids by simple, fourth generation methods, Pure Appl. Chem. 51(1979) 689-703. |
| [5] | Y. Shi, J.T. Wilmot, L.U. Nordstrom, et al., Total synthesis, relay synthesis, and structural confirmation of the C18-norditerpenoid alkaloid neofinaconitine. J. Am. Chem. Soc. 135 (2013) 14313–14320. DOI:10.1021/ja4064958 |
| [6] | C.J. Marth, G.M. Gallego, J.C. Lee, et al., Network-analysis-guided synthesis of weisaconitine D and liljestrandinine. Nature 528 (2015) 493–498. DOI:10.1038/nature16440 |
| [7] | Y. Nishiyama, S. Yokoshima, T. Fukuyama. Total synthesis of (-)-cardiopetaline. Org. Lett. 18 (2016) 2359–2362. DOI:10.1021/acs.orglett.6b00789 |
| [8] |
(a) J. L. van der Baan, J. W. F. K. Barnick, G. van Beek, et al. , Total synthesis of C19-diterpene alkaloids: construction of a functionalized ABCD-ring system, Tetrahedron 48(1992) 2773-2784; (b) L. C. Baillie, J. R. Bearder, W. S. Li, et al. , Studies into the synthesis of a sub-unit of the neurotoxic alkaloid methyllycaconitine, J. Chem. Soc. Perkin Trans. 1(1998) 4047-4055; (c) D. F. Taber, J. L. Liang, B. Chen, et al. , A model study toward the total synthesis of N-deacetyllappaconitine, J. Org. Chem. 70(2005) 8739-8742; (d) G. A. Kraus, S. Kesavan, Preparation of advanced intermediates for the synthesis of both methyllycaconitine and racemulsonine via a common intermediate, Tetrahedron Lett. 46(2005) 1111-1113; (e) R. M. Conrad, J. Du Bois, C-H amination in synthesis: an approach to the assembly of the B/C/D ring system of aconitine, Org. Lett. 9(2007) 5465-5468; (f) K. Hagiwara, T. Tabuchi, D. Urabea, et al. , Expeditious synthesis of the fused hexacycle of puberuline C via a radical-based cyclization/translocation/cyclization process, Chem. Sci. 7(2016) 4372-4378; (g) T. Tabuchi, D. Urabe, M. Inoue, Construction of the fused pentacycle of talatisamine via a combination of radical and cationic cyclizations, J. Org. Chem. 81(2016) 10204-10213. |
| [9] |
(a) Z. G. Liu, L. Xu, Q. H. Chen, et al. , Construction of A/E/F ring systems of C19-diterpenoid alkaloids with both C-1 and C-6 oxygen functions, Tetrahedron 68(2012) 159-165; (b) H. Cheng, L. Xu, D. L. Chen, et al. , Construction of functionalized B/C/D ring system of C19-diterpenoid alkaloids via intramolecular Diels-Alder reaction followed by Wagnere-Meerwein rearrangement, Tetrahedron 68(2012) 1171-1176; (c) Z. G. Liu, H. Cheng, M. J. Ge, et al. , PIDA-promoted intramolecular transannular aziridination to synthesize bridged azatricyclic amines related to methyllycaconitine, Tetrahedron 69(2013) 5431-5437; (d) R. H. Mei, Z. G. Liu, H. Cheng, et al. , Synthesis of the 10-azatricyclo[3. 3. 2. 04, 8]decan core of C20-diterpenoid alkaloid racemulsonine via iodine(Ⅲ) promoted transannular aziridination reaction, Org. Lett. 15(2013) 2206-2209; (e) H. Cheng, F. H. Zeng, D. Ma, et al. , Expedient construction of the ABEF azatetracyclic ring systems of lycoctonine-type and 7, 17-seco-type C19-diterpenoid alkaloids, Org. Lett. 16(2014) 2299-2301; (f) M. L. Jiang, Y. J. Meng, W. Y. Xiong, et al. , Construction of functionalized ABEF ring system of C20-diterpenoid alkaloid racemulosine, Tetrahedron Lett. 57(2016) 1610-1612; (g) Y. L. Li, M. C. Liu, Y. J. Meng, Two new entries to the ABF tricyclic ring system of 7, 17-seco-type C19-diterpenoid alkaloids via free radical cyclization and[3+2] cycloaddition of nitrile oxide, Tetrahedron 72(2016) 3171-3176. |
| [10] |
(a)J. Marco-Contelles, B. Sánchez, Stereoelectronic effects in the 6-exo free radical cyclization of acyclic sugar derivatives: synthesis of branched chain cyclitols, J. Org. Chem. 58(1993) 4293-4297; (b) D. Batty, D. Crich, S. M. Fortt, Synthesis of a 1a, 25-dihydroxyvitamin D3A ring model by an acyl radical cyclization, J. Chem. Soc. Chem. Commun. (1989) 1366-1368; (c) J. Quirante, C. Escolano, F. Diaba, et al. , Radical promoted cyclizations of trichloroacetamides with silyl enol ethers and enol acetates: the role of the hydride reagent[tris(trimethylsilyl)silane vs. tributylstannane], J. Chem. Soc. Perkin Trans. 1(1999) 1157-1162; (d) D. J. Wardrop, W. Zhang, N-methoxy-N-acylnitrenium ions: Application to the formal synthesis of (±)-desmethylamino FR901483, Org. Lett. 3(2001) 2353-2356. |
| [11] |
(a) M. H. Filippini, R. Faure, J. Rodriguez, One-pot base-promoted tandem michael addition-intramolecular aldolization. Stereoselective synthesis and reactivity of 2-hydroxybicyclo[3. 2. 1]octan-8-ones, J. Org. Chem. 60(1995) 6872-6882; (b) H. Hagiwara, M. Fukushima, K. Kinugawa, et al. , First total syntheses of bicyclic marine sesquiterpenoids drechslerines A and B, Tetrahedron 67(2011) 4061-4068. |
| [12] | C. Petrier, J.L. Luche. Allylzinc reagents additions in aqueous media. J. Org. Chem. 50 (1985) 910–912. DOI:10.1021/jo00206a047 |
| [13] |
(a) Y. K. Chen, R. K. Peddinti, C. C. Liao, Diastereoselective intramolecular Diels-Alder reactions of masked o-benzoquinones: a short entry to highly functionalized tricyclic[m. 2. 2. 0] ring systems, Chem. Commun. (2001) 1340-1341; (b) S. K. Chittimalla, H. Y. Shiao, C. C. Liao, Domino retro Diels-Alder/Diels-Alder reaction: anefficient protocolfor the synthesis of highlyfunctionalized bicyclo[2. 2. 2]octenones and bicyclo[2. 2. 2]octadienones, Org. Biomol. Chem. 4(2006) 2267-2277; (c) H. Cheng, F. H. Zeng, X. Yang, et al. , Collective total syntheses of atisane-type diterpenes and atisine-type diterpenoid alkaloids: (±)-spiramilactone B, (±)-spiraminol, (±)-dihydroajaconine, and (±)-spiramines C and D, Angew. Chem. Int. Ed. 55(2016) 392-396. |