 2017, Vol. 28
2017, Vol. 28 



b Division of Advanced Nanomaterials, Suzhou Institute of Nano-tech and Nano-bionics, Chinese Academy of Sciences, Suzhou 215125, China;
c University of Chinese Academy of Sciences, Beijing 100049, China;
d University of Science and Technology of China, Hefei 230026, China
Formic acid (FA) is a promising liquid for hydrogen storage and generation because it is nontoxic under normal conditions, available from biomass processing or reduction of carbon dioxide, and has remarkable hydrogen content (4.4 wt%) [1-3]. The hydrogen from FA is suitable for proton exchange membrane fuel cell (PEMFC), because the gas evolved from FA has trace or no CO [3-5]. Hence, this technology has aroused growing interests for FA dehydrogenation, and many catalysts have been developed for the FA dehydrogenation reaction [6-14].
The catalysts for FA dehydrogenation can be classified into two main categories including homogeneous catalysts [4, 7, 10, 15-19] and heterogeneous catalysts [20-29], which have their own unique advantages. For example, the homogeneous catalysts based on Ir, Ru, Rh and Fe organometallic complex usually have high selectivity and catalytic activity, while the heterogeneous catalysts based on Pt, Au, Pd, Ag, Co nanoparticles and nano-alloy can be easily separated, controlled and recycled.
Some nanocatalysts have been designed for the FA dehydrogenation since 2008 [30-35]. However, there are still some difficulties for these catalysts to be practically applied. For example, the evolved gas from FA dehydrogenation by some nanocatalysts contains trace CO, which could occupy the catalytic active site, and lower down the performance of PEMFC. PEMFCs are sensitive to the trace CO and the hydrogen generating rate, just a little CO would poison the catalyst in the PEMFC. The unstable hydrogen generating rate would also lead to the fluctuation of generated current, which could damage the electricity equipment. Therefore, developing a stable catalyst with high selectivity for FA dehydrogenation is significant.
In this work, we designed a self-supporting fuel cell system, in which H2 from FA is supplied into the fuel cell, and the exhaust heat from the fuel cell supported the FA dehydrogenation. To realize the system, we synthesized a highly active and selective IrCp*Cl2bpym for FA dehydrogenation in this research. The catalyst shows excellent catalytic performance for FA dehydrogenation. The turnover frequency (TOF) was up to 144, 000 h-1 at 90 ℃, and 7150 h-1 at 50 ℃ at mild conditions. In addition, no CO was detected in the evolved gas. Therefore, the evolved gas could be applied in the PEMFC directly, generating current continuously.
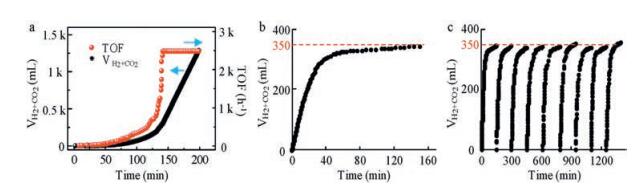
2. Results and discussion 2.1. Catalytic performance for FA decompositionThe IrCp*Cl2bpym has excellent catalytic activity for FA dehydrogenation, but the catalyst can only exhibit its high catalytic activity after experiencing a catalyst activation process. Fig. 1a exhibits the gas generation after the catalyst being added in the FA solution. The gas generation rate was very slow at the beginning of FA dehydrogenation, but kept accelerating during the initial 150 minutes. This was the activation process of the catalyst IrCp*Cl2bpym. Then the catalyst would keep stable and high activity for the FA dehydrogenation, and the TOF value was up to 2490 h-1 at 40 ℃.

|
Download:
|
| Fig. 1. Gas generation from FA dehydrogenation. (a) Gas generation process from FA dehydrogenation in the FA-SF mixture solution at 40 ℃. (b) Gas generation from 300 mL FA in 4 mL water at 50 ℃. (c) Cycles test of the FA dehydrogenation at 50 ℃. Condition: catalyst (4.7 mg) | |
{kind=link}
Fig. 1b shows the volume of the generating gas during the FA dehydrogenation. The gas generating rate was very high initially and then decreased slowly during the FA dehydrogenation. This could be attributed to the sharp decrease of FA concentration, because the concentration of FA could exert tremendous influence on the FA dehydrogenation. When we added another 300 mL FA in the same reaction solution, the high catalytic activity could recover quickly (Fig. 1c). Fig. 1c shows that the catalyst could keep the stable and high catalytic activity for the FA dehydrogenation after several cycles. 350 mL gas would be generated from 300 mL FA during 150 minutes. The conversion ratio of FA was up to 93.2% and no carbon monoxide was detected in the evolved gas (Supporting information). The high conversion ratio and selectivity could ensure the hydrogen from FA decomposition to be applied in the PEMFC directly.
In our previous works, we found the FA concentration and the proportion of sodium formate (SF) to FA had great influence on the FA dehydrogenation [32]. Therefore, we also focused on studying the influence of FA concentration and proportion of SF to FA in this research. Fig. 2a exhibits the influence of FA concentration on FA dehydrogenation. As the concentration was lower than 4 mol/L, the catalytic activity would increase with the FA concentration. The TOF value would achieve 230 h-1 when the FA concentration was 4 mol/L. Then the catalytic activity would decrease slightly when the concentration was over 4 mol/L. The catalytic activity was more sensitive to low FA concentration, and could keep a relatively stable catalytic activity for the dehydrogenation of high-concentration FA. This means that the hydrogen would be generated stably if the IrCp*Cl2bpym was applied for high concentration of FA, and this was a key step to generating stable current from PEMFCs.

|
Download:
|
| Fig. 2. Effect of FA concentration and the proportion of sodium formate (SF) to FA on the catalytic activity of IrCp*Cl2bpym for FA dehydrogenation. (a) FA concentration dependence of FA dehydrogenation at 30 ℃. (b) Proportion of SF to FA dependence of FA dehydrogenation at 30 ℃, [FA] + [SF] = 6 mol/L | |
{kind=link}
Fig. 2b shows the TOF values obtained in the FA-SF mixture solutions at different proportions of SF to FA. The proportions had great influence on the catalytic activity for hydrogen generation from FA dehydrogenation. The TOF would rise nearly threefold to 655 h-1 at the appropriate proportion of SF to FA. The appropriate addition of SF could accelerate the FA dehydrogenation rate. The highest TOF value was obtained when the proportion of SF to FA was 0.5. Higher or lower proportion would decrease the catalytic rate sharply when the total concentration of FA and SF was 6 mol/L.
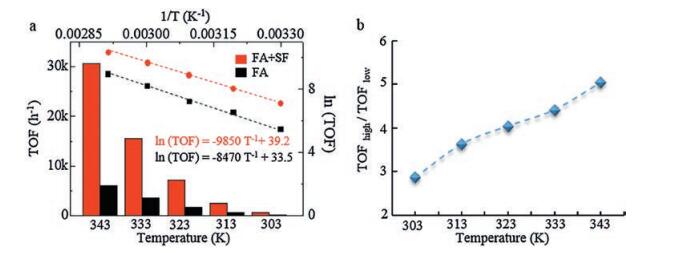
The SF could accelerate the catalytic rate. Therefore, we further studied its influence on FA dehydrogenation at different temperatures. Fig. 3a shows the TOF values of FA dehydrogenation in FA or FA-SF solution at different temperatures. The FA dehydrogenation rate increased with temperature in both FA and FA-SF solutions. The TOF values for FA and FA-SF solutions were 230 h-1 and 655 h-1 at 30 ℃, and up to 29300 h-1 and 5820 h-1 at 70 ℃, respectively. The gas generation rate in FA solution was obviously lower than that in FA-SF solution. The higher temperature would widen this gap of TOFs in FA-SF and FA solution. For example, the TOF ratio was 2.86 at 30 ℃, while the ratio would increase to 5.04 at 70 ℃ (Fig. 3b). That means the TOF value in FA-SF solution was 5.04 times higher than that in FA solution, and the SF would greatly increase the reaction rate at higher temperature. The SF may accelerate some key steps during the FA dehydrogenation, which we would discuss in the following part.

|
Download:
|
| Fig. 3. Temperature dependence of FA dehydrogenation. (a) Arrhenius plot and TOF values of FA decomposition at different temperatures. Black line condition: FA 4 mol/L. Red line condition: [FA] + [SF] = 6 mol/L, [SF]/[FA] = 0.5. (b) TOF ratio under different conditions at same temperature in a | |
{kind=link}
We could gain Arrhenius equations with temperature dependence of the TOF value. The estimated apparent activation energy (Ea) for FA dehydrogenation could be calculated through the two Arrhenius equations (Fig. 3a). The Ea was 81.9 kJ/mol in FA-SF solution and was 70.4 kJ/mol in FA solution. The different Ea values revealed that this catalyst exhibited different activities for FA dehydrogenation in water or FA-SF solution intrinsically. Hence, we proposed a possible reaction mechanism for the FA dehydrogenation in water or FA-SF solution based on the results discussed above (Fig. 4).

|
Download:
|
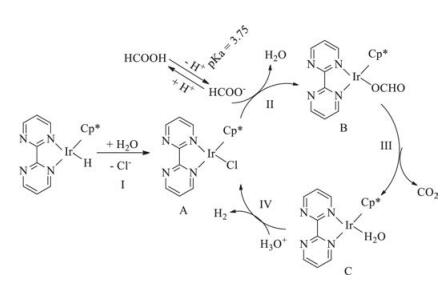
| Fig. 4. Proposed mechanism for FA dehydrogenation | |
{kind=link}
2.2. Catalytic mechanism for FA decomposition
The catalyst dissolved in water and formed water-coordinated iridium complex (A), which was the beginning of the catalytic cycle. The HCOO- coordinated to the complex A, and formed the formato complex B subsequently (Step Ⅱ). The HCOO- was derived from the ionization of FA. As an organic acid, the FA could not completely ionize in water. Therefore, adding formate salt could contribute to raising the HCOO- concentration in water, and accelerating the reaction rate of Step Ⅱ. This well explained the role of SF in FA dehydrogenation. The iridium hydride complex C formed after the release of CO2 through decarboxylation (Step Ⅲ). At last, the iridium hydride complex could react with H3O+ in water, generating H2 and water-coordinated iridium complex (A). Then the total catalytic cycle finished (Step Ⅳ). The Step Ⅳ was a direct step for H2 generation, which was affected by the H3O+ concentration. Although adding SF could raise the HCOO- concentration in water and favor the Step Ⅱ, the H3O+ concentration would decrease for the alkalinity of SF. Consequently, excessive SF in water could go against the H2 generation (Step Ⅳ), which had been confirmed in Fig. 2b.
2.3. FA decomposition applied to the PEMFCThe hydrogen from FA decomposition could be applied in PEMFCs for current generation. The products of FA dehydrogenation were hydrogen and carbon dioxide, and no carbon monoxide was detected from the evolved gas. Therefore, the evolved gas had no damage to the catalyst in the PEMFC, and could be applied in the PEMFC directly. As showed in Fig. 5, the evolved gas from FA dehydrogenation could generate current directly through an air breathing PEMFC with 2 cm2 working area. The power density of fuel cell using this gas could achieve 64 mW/cm2 when the gas flow rate was 30 mL/min at 20 ℃. When the same membrane electrode assembly (MEA) used pure hydrogen as fuel, the power density was 75 mW/cm2. The values of power density of fuel cell using different fuel were almost the same if the fuel cell discharged current at higher current potential or lower current density. Compared with the pure hydrogen, the hydrogen evolved from FA was much more convenient and safe for transportation and storage. The FA could show its great potential for hydrogen storage if this catalyst was applied and made the mobile hydrogen generation come true.

|
Download:
|
| Fig. 5. Current generation by H2 from FA dehydrogenation. Polarization curves of 2 cm2 membrane electrode assembly (MEA) with different fuels. Anode and cathode: Pt loading (0.5 mg/cm2), gas rate from FA dehydrogenation (30 mL/min), pure hydrogen rate 15 mL/min | |
{kind=link}
In this work, we focus on the development of catalyst and the application of hydrogen in single fuel cell. In future work, we will integrate the hydrogen production part into the fuel cell system to make a self-supporting fuel cell system. In the system, H2 from FA is supplied into fuel cell, and the exhaust heat from fuel cell supports the FA dehydrogenation.
3. ConclusionA homogeneous catalyst IrCp*Cl2bpym for FA dehydrogenation is reported in this work. The catalyst shows high catalytic activity and selectivity for hydrogen generation from FA. The TOF is as high as 7150 h-1 at 50 ℃, and is up to 144000 h-1 at 90 ℃. In addition, the catalyst can keep a stable catalytic activity for FA dehydrogenation after several cycles of test, and the conversion ratio of FA would achieve 93.2%, no carbon monoxide is detected in the evolved gas. We furthermore study the influence of FA concentration and the proportion of SF to FA on FA dehydrogenation, and propose possible mechanism based on these results. According to the high catalytic activity and selectivity of the IrCp*Cl2bpym for FA dehydrogenation, the evolved hydrogen could be applied in the PEMFC directly. The power density driven by evolved hydrogen could compare favorably with that driven by pure hydrogen. Attributing to its convenient transportation and safer storage, the hydrogen from FA dehydrogenation has great potential for the current generation in the mobile device.
4. Experimental 4.1. Chemicals and materialsAll chemicals were commercial and used without further purification unless specified. Chloroiridic acid (H2IrCl6·6H2O, Shanghai Tuosi Chemical Co., Ltd, Ir wt% > 35%), formic acid (HCOOH, Sinopharm Chemical Reagent. Co., Ltd. >98%), ethanol (C2H5OH, Sinopharm Chemical Reagent, Co., Ltd, >99.7%), 1, 2, 3, 4, 5-pentamethylcyclopentadiene (C10H16, Sun Chemical Technology (Shanghai) Co., Ltd, 97%), 1, 10-phenanthroline (C12H8N2, Sinopharm Chemical Reagent, Co., Ltd. >99%), 2, 2'-bipyrimidyl(C8H6N4, China Langchem Co. Ltd, >98%), sodium hydroxide (NaOH, Sinopharm Chemical Reagent, Co., Ltd, >96%), ether (C4H10O, Sinopharm Chemical Reagent, Co., Ltd, >99.5%), ultrapure water was prepared by Thermo PureLab Ultra Genetic. The 1H NMR data was collected by Varian 400 M.
4.2. Synthesis of [IrCp*Cl2]2Excess 1, 2, 3, 4, 5-pentamethylcyclopentadiene was added into H2IrCl6 methanol solution. The molar ratio of 1, 2, 3, 4, 5-pentamethylcyclopentadiene to H2IrCl6 was about 2.5:1. The mixture was stirred under reflux for 37 h, and then cooled to 0 ℃. The yellow brown product will be gained after filtration and washed with ether.
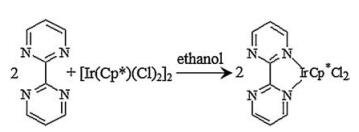
4.3. Synthesis of IrCp*Cl2bpym200 mg [IrCp*Cl2]2 Ethanol solution was treated with 79 mg 2, 2'-bipyrimidyl in a 25 mL round-bottomed flask. The mixture solution was stirred and heated under reflux for 8 hours. The insoluble [IrCp*Cl2]2 would dissolve and the solution turned pale yellow during the reflux. Yellow product could be gained after removing the ethanol by rotary distillation under reduced pressure. Synthesis process refer to literature partially (Scheme 1) [36]. The IrCp*Cl2bpym could also been gained by ligand transfer between [IrCp*Cl2]2bpym and 1, 10-phenanthroline (Supporting information). [IrCp*Cl2]2bpym was also an outstanding catalyst for FA dehydrogenation [17].

|
Download:
|
| Scheme1. Schematic illustration for IrCp*Cl2bpym synthesis | |
{kind=link}
4.4. FA dehydrogenated by IrCp*Cl2bpym
First, a one-necked round-bottomed flask (25 mL) containing 10 mL FA solution was kept at the preset temperature (20-90 ℃) in a water bath under ambient atmosphere. The measurement started as soon as the catalyst was added into the solution. A graduated buret filled with water or a 100 mL syringe was connected to the reaction flask to measure the volume of evolved gas from FA dehydrogenation. The volume change was recorded by a digital camera. The temperature for volume measurement was kept at 20 ℃ during the measurements. The evolved gas would be analyzed by gas chromatography GC-G5 (Beijing Persee General Instrument Co., Ltd), the gas chromatography assembled with TDX-1 column, FID, TCD and methanizer, N2 as carrier gas.
4.5. TOF calculationThe TOF calculation here is based on the number of Ir atoms in catalyst. The calculation equation is

|
Where Patm is the atmospheric pressure (101325 Pa), Vrate is the gas generating rate (m3/h), ncata is the total mole number of IrCp*Cl2bpym in the reaction solution, R is the universal gas constant (8.314 m3 Pa/mol/K), and T (293.15 K) is the temperature of laboratory. Formic acid decomposes through the following chemical equation, HCOOH → H2 + CO2. 1 mol formic acid will generate 1 mol gas (H2 + CO2).
AcknowledgmentThe authors are grateful for financial support granted by Ministry of Science and Technology of China (Nos. 2016YFE0105700, 2016YFA0200700), the National Natural Science Foundation of China (Nos. 21373264, 21573275), the Natural Science Foundation of Jiangsu Province (No. BK20150362), Suzhou Institute of Nano-tech and Nano-bionics (No. Y3AAA11004) and Thousand Youth Talents Plan (No. Y3BQA11001).
| [1] | Q.L. Zhu, Q. Xu. Liquid organic and inorganic chemical hydrides for highcapacity hydrogen storage. Energy Environ. Sci. 8 (2015) 478–512. DOI:10.1039/C4EE03690E |
| [2] | M. Yadav, Q. Xu. Liquid-phase chemical hydrogen storage materials. Energy Environ. Sci. 5 (2012) 9698–9725. DOI:10.1039/c2ee22937d |
| [3] | M. Grasemann, G. Laurenczy. Formic acid as a hydrogen source-recent developments and future trends. Energy Environ. Sci. 5 (2012) 8171–8181. DOI:10.1039/c2ee21928j |
| [4] | C. Fellay, P.J. Dyson, G. Laurenczy. A viable hydrogen-storage system based on selective formic acid decomposition with a ruthenium catalyst. Angew. Chem. Int. Ed. 120 (2008) 4030–4032. DOI:10.1002/(ISSN)1521-3757 |
| [5] | T.C. Johnson, D.J. Morris, M. Wills. Hydrogen generation from formic acid and alcohols using homogeneous catalysts. Chem. Soc. Rev. 39 (2010) 81–88. DOI:10.1039/B904495G |
| [6] | X. Gu, Z.H. Lu, H.L. Jiang, T. Akita, Q. Xu. Synergistic catalysis of metal-organic framework-immobilized Au-Pd nanoparticles in dehydrogenation of formic acid for chemical hydrogen storage. J. Am. Chem. Soc. 133 (2011) 11822–11825. DOI:10.1021/ja200122f |
| [7] | S. Fukuzumi, T. Kobayashi, T. Suenobu. Efficient catalytic decomposition of formic acid for the selective generation of H2 and H/D exchange with a watersoluble rhodium complex in aqueous solution. ChemSusChem 1 (2008) 827–834. DOI:10.1002/cssc.v1:10 |
| [8] | Y. Himeda. Highly efficient hydrogen evolution by decomposition of formic acid using an iridium catalyst with 4, 4'-dihydroxy-2, 2'-bipyridine. Green Chem. 11 (2009) 2018–2022. DOI:10.1039/b914442k |
| [9] | A.K. Singh, S. Jang, J.Y. Kim, et al., One-pot defunctionalization of lignin-derived compounds by dual-functional Pd50Ag50/Fe3O4/N-rGO Catalyst. ACS Catal. 5 (2015) 6964–6972. DOI:10.1021/acscatal.5b01319 |
| [10] | P. Sponholz, D. Mellmann, H. Junge, M. Beller. Towards a practical setup for hydrogen production from formic acid. ChemSusChem 6 (2013) 1172–1176. DOI:10.1002/cssc.201300186 |
| [11] | K. Tedsree, T. Li, S. Jones, et al., Hydrogen production from formic acid decomposition at room temperature using a Ag-Pd core-shell nanocatalyst. Nat. Nanotechnol. 6 (2011) 302–307. DOI:10.1038/nnano.2011.42 |
| [12] | S. Wu, F. Yang, H. Wang, et al., Mg2+-assisted low temperature reduction of alloyed AuPd/C:an efficient catalyst for hydrogen generation from formic acid at room temperature. Chem. Commun. 51 (2015) 10887–10890. DOI:10.1039/C5CC02604K |
| [13] | J.M. Yan, Z.L. Wang, L. Gu, et al., AuPd-MnOx/MOF-Graphene:An Efficient Catalyst for Hydrogen Production from Formic Acid at Room Temperature. Adv.Energy Mater. 5 (2015) 1500107. DOI:10.1002/aenm.201500107 |
| [14] | S. Zhang, O. Metin, D. Su, S. Sun. Monodisperse AgPd alloy nanoparticles and their superior catalysis for the dehydrogenation of formic acid. Angew. Chem. Int. Ed. 52 (2013) 3681–3684. DOI:10.1002/anie.201300276 |
| [15] | A.V. Bavykina, M.G. Goesten, F. Kapteijn, M. Makkee, J. Gascon. Efficient production of hydrogen from formic acid using a covalent triazine framework supported molecular catalyst. ChemSusChem 8 (2015) 809–812. DOI:10.1002/cssc.v8.5 |
| [16] | Z. Wang, S.M. Lu, J. Li, J. Wang, C. Li. Unprecedentedly high formic acid dehydrogenation activity on an iridium complex with an N, N'-diimine ligand in water. Chem.-Eur. J. 21 (2015) 12592–12595. DOI:10.1002/chem.201502086 |
| [17] | J.F. Hull, Y. Himeda, W.H. Wang, et al., Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 4 (2012) 383–388. DOI:10.1038/nchem.1295 |
| [18] | A. Boddien, F. Gartner, R. Jackstell, et al., ortho-Metalation of iron(0) tribenzylphosphine complexes:homogeneous catalysts for the generation of hydrogen from formic acid. Angew. Chem. Int. Ed. 49 (2010) 8993–8996. DOI:10.1002/anie.v49.47 |
| [19] | S. Fukuzumi, T. Kobayashi, T. Suenobu. Unusually large tunneling effect on highly efficient generation of hydrogen and hydrogen isotopes in pH-selective decomposition of formic acid catalyzed by a heterodinuclear iridiumruthenium complex in water. J. Am. Chem. Soc. 132 (2010) 1496–1497. DOI:10.1021/ja910349w |
| [20] | K. Jiang, K. Xu, S. Zou, W.B. Cai. B-doped Pd catalyst:boosting roomtemperature hydrogen production from formic acid-formate solutions. J. Am. Chem. Soc. 136 (2014) 4861–4864. DOI:10.1021/ja5008917 |
| [21] | Y. Chen, Q.L. Zhu, N. Tsumori, Q. Xu. Immobilizing highly catalytically active noble metal nanoparticles on reduced graphene oxide:a non-noble metal sacrificial approach. J. Am. Chem. Soc. 137 (2015) 106–109. DOI:10.1021/ja511511q |
| [22] | Y.Y. Cai, X.H. Li, Y.N. Zhang, et al., Highly efficient dehydrogenation of formic acid over a palladium-nanoparticle-based Mott-Schottky photocatalyst. Angew. Chem. Int. Ed. 52 (2013) 11822–11825. DOI:10.1002/anie.v52.45 |
| [23] | Q.Y. Bi, X.L. Du, Y.M. Liu, et al., Efficient subnanometric gold-catalyzed hydrogen generation via formic acid decomposition under ambient conditions. J. Am. Chem. Soc. 134 (2012) 8926–8933. DOI:10.1021/ja301696e |
| [24] | Z.L. Wang, J.M. Yan, Y. Ping, et al., An efficient CoAuPd/C catalyst for hydrogen generation from formic acid at room temperature. Angew. Chem. Int. Ed. 52 (2013) 4406–4409. DOI:10.1002/anie.201301009 |
| [25] | L. Yang, X. Hua, J. Su, et al., Highly efficient hydrogen generation from formic acid-sodium formate over monodisperse AgPd nanoparticles at room temperature. App. Catal. B:Environ. 168-169 (2015) 423–428. DOI:10.1016/j.apcatb.2015.01.003 |
| [26] | J.S. Yoo, Z.J. Zhao, J.K. Nørskov, F. Studt. Effect of Boron Modifications of Palladium Catalysts for the Production of Hydrogen from Formic Acid. ACS Catal. 5 (2015) 6579–6586. DOI:10.1021/acscatal.5b01497 |
| [27] | Y.L. Qin, Y.C. Liu, F. Liang, L.M. Wang. Preparation of Pd-Co-based nanocatalysts and their superior applications in formic acid decomposition and methanol oxidation. ChemSusChem 8 (2015) 260–263. DOI:10.1002/cssc.201402926 |
| [28] | F.F. Li, J.N. Gu, X.C. Zhou. Single molecule electro-catalysis of non-fluorescent molecule. Chin. Chem. Lett. 26 (2015) 1514–1517. DOI:10.1016/j.cclet.2015.09.013 |
| [29] | Y.X. Wang, T.H. Chen. A high dispersed Pt0.35Pd0.35Co0.30/C as superior catalyst for methanol and formic acid electro-oxidation. Chin. Chem. Lett. 25 (2014) 907–911. DOI:10.1016/j.cclet.2014.04.031 |
| [30] | X. Zhou, Y. Huang, W. Xing, et al., High-quality hydrogen from the catalyzed decomposition of formic acid by Pd-Au/C and Pd-Ag/C. Chem. Commun. (2008) 3540–3542. |
| [31] | Y. Huang, X. Zhou, M. Yin, C. Liu, W. Xing. Novel PdAu@Au/C Core-Shell Catalyst:Superior Activity and Selectivity in Formic Acid Decomposition for Hydrogen Generation. Chem. Mater. 22 (2010) 5122–5128. DOI:10.1021/cm101285f |
| [32] | X. Zhou, Y. Huang, C. Liu, et al., Available hydrogen from formic acid decomposed by rare earth elements promoted Pd-Au/C catalysts at low temperature. ChemSusChem 3 (2010) 1379–1382. DOI:10.1002/cssc.201000199 |
| [33] | W. Wang, T. He, X. Liu, et al., Highly active carbon supported Pd-Ag nanofacets catalysts for hydrogen production from HCOOH. ACS Appl. Mater. Interfaces 8 (2016) 20839–20848. DOI:10.1021/acsami.6b08091 |
| [34] | M.J. Ren, Y. Zhou, F.F. Tao, et al., Controllable modification of the electronic structure of carbon-supported core-shell Cu@Pd catalysts for formic acid oxidation. J. Phys. Chem. C 118 (2014) 12669–12675. DOI:10.1021/jp5033417 |
| [35] | M. Ren, Y. Kang, W. He, et al., Origin of performance degradation of palladiumbased direct formic acid fuel cells. App. Catal. B-Environ. 104 (2011) 49–53. DOI:10.1016/j.apcatb.2011.02.029 |
| [36] | P. Govindaswamy, J. Canivet, B. Therrien, et al., Mono and dinuclear rhodium, iridium and ruthenium complexes containing chelating 2, 20-bipyrimidine ligands:Synthesis, molecular structure, electrochemistry and catalytic properties. J. Organomet. Chem. 692 (2007) 3664–3675. DOI:10.1016/j.jorganchem.2007.04.048 |