 2017, Vol. 28
2017, Vol. 28 


 ,
Hu-Ri Piaoa
,
Hu-Ri Piaoa
b Department of Pharmacy, Jilin Medical University, Jilin 132013, China
The management of bacterial infections has become increasingly difficult with the emergence of antimicrobial resistance. In particular, the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) are causing significant problems worldwide [1]. Methicillin-resistant S. aureus (MRSA) is a major pathogen causing both hospital-and community-acquired infections. Among these organisms, MRSA alone accounts for nearly one-half of the deaths attributed to antibiotic-resistant infections. In 2013, the Centers for Disease Control and Prevention reported that more than 11, 000 people died from MRSA-related infections in the United States alone [2]. Therefore, there is an urgent need for the development of new antibacterial agents with divergent and unique structures and mechanisms of action that differ from those of existing antimicrobial agents [3].
Medicinal chemistry studies have shown that a 1, 3, 5-triazine scaffold is an outstanding pharmacophore, and many molecules with this scaffold have been reported as antibacterial agents. 1, 2-Dihydro-1, 3, 5-triazine (Baker triazines) compounds are by far the best known, with several studies exploring their potential as inhibitors of eukaryotic dihydrofolate reductase (DHFR), an important enzyme in the de novo pathway of purine and thymidine synthesis, and small molecules targeting this enzyme have demonstrated utility as potential antibiotics [4, 5]. Feng et al. reported that the compound N2-methyl-6-(5-methylisoxazol-3-yloxy)-N4-(4-(trifluoromethyl)phenyl)-1, 3, 5-triazine-2, 4-diamine displayed an antimicrobial effect as high as 97.7% at a concentration of 200 mg/mL [6]. Singga et al. found that triazine derivatives inhibited some Gram-positive and Gram-negative bacteria by inhibiting cell membrane growth [7].
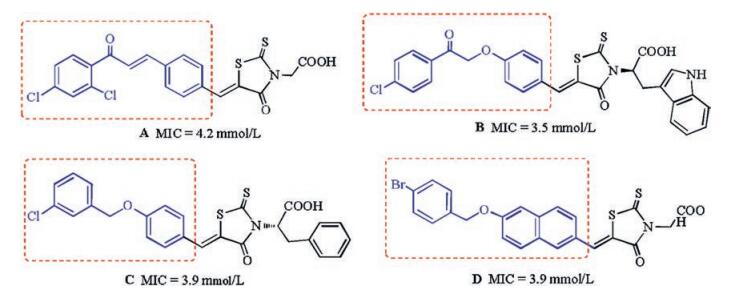
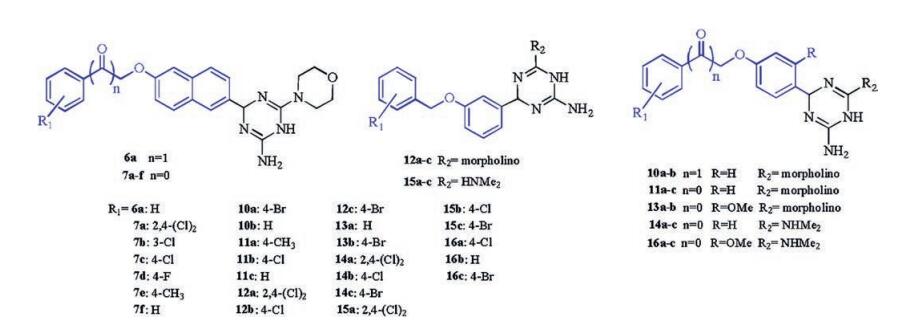
In our previous work, we found that several rhodanine derivatives bearing chalcone [8], (2-oxo-2-phenylethoxy)benzylidene [9], and (benzyloxy)benzylidene [10] moieties (Fig. 1) showed moderate to strong activities against several Grampositive bacterial strains, including multidrug-resistant clinical isolates. Unfortunately, compounds belonging to this series did not show any bacteriostatic activity against Gram-negative bacteria. As a part of our ongoing studies toward the development of novel antibacterial agents, here we designed and synthesized a series of 1, 4-dihydro-1, 3, 5-triazine derivatives using a hybrid strategy, in which the rhodamine moiety was replaced by a dihydrogen triazine scaffold. Thus, a series of 26 novel 1, 4-dihydro-1, 3, 5-triazine derivatives (Fig. 2) were synthesized and evaluated for their antibacterial activities in vitro. The substituents on the phenyl ring were changed simultaneously to investigate their contribution to the biological activity.

|
Download:
|
| Fig. 1. Previously reported antibacterial compounds. | |

|
Download:
|
| Fig. 2. Synthesized target compounds. | |
2. Results and discussion
The in vitro antibacterial activities of the synthesized compounds were evaluated using a 96-well microtiter plate and a serial dilution method to obtain the minimum inhibitory concentrations (MICs) for different strains (including multidrug-resistant clinical isolates). Two Gram-positive strain (S. aureus RN 4220 and S. Mutans KTCT 3289), five Gram-negative strains (Escherichia coli KCTC 1924, E. coli CCARM 1356, P. aeruginosa 2742, P. aeruginosa 2004, and Salmonella enterica serovar Typhimurium 2421) and one fungus (Candida albicans 7535) were used for the biological assay. Gatifloxacin, moxifloxacin, norfloxacin, and oxacillin were used as positive controls for antibacterial activity, and fluconazole and itraconazole were used as positive controls for antifungal activity.
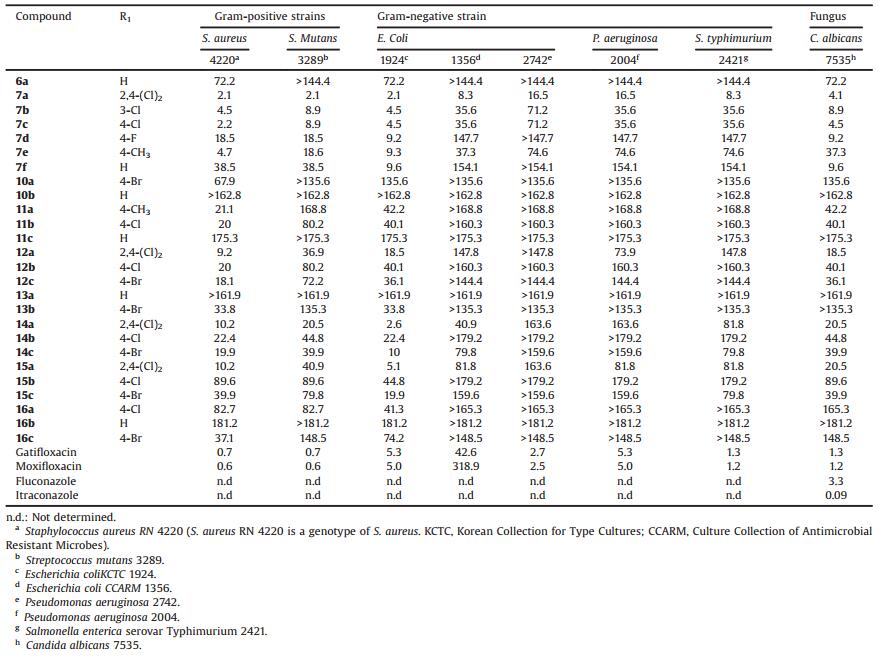
The in vitro antibacterial and antifungal activities of the synthesized compounds are shown in Table 1. Most of the synthesized compounds showed potent inhibitory activities against the different bacteria and one fungus, with MICs ranging from 2.1 to 181.2 mmol/L. Compounds 7a and 7c exhibited the highest activity of all the compounds synthesized against S. aureus 4220, with an MIC of 2.1 mmol/L. Against the Gram-negative E. coli 1924, compounds 7a, 7b, 7c, 14a, and 15a were equipotent or more potent than the positive controls gatifloxacin and moxifloxacin (MIC = 5.0 mmol/L), with an MIC of 2.1 mmol/L. The potency of compound 7a, with an MIC of 8.3 mmol/L, was 5-fold greater than that of the positive control gatifloxacin (MIC = 42.6 mmol/L). The potency of compounds 7b, 7c, 7e, and 14a, with an MIC of 35.6mmol/L against E. coli 1356, was 9-fold greater than that of the positive control moxifloxacin (MIC = 318.9 mmol/L). However, for the P. aeruginosa 2742 and 2004 strains, only compound 7a exhibited good activity, with an MIC of 16.5 mmol/L. Against C. albicans 7535, compounds 7a and 7c displayed the strongest potency of all the compounds synthesized, with an MIC of 4.1 mmol/L, which was slightly lower than that of fluconazole (MIC = 3.3 mmol/L). Previously, we reported that rhodamine derivatives bearing benzyloxy benzaldehyde or benzyloxy naphthaldehyde moieties did not show any activity against the Gramnegative bacteria E. coli and P. aeruginosa [11], but dihydro-1, 3, 5-triazine derivatives displayed moderate to high levels of inhibition against these strains, indicating that the dihydro-1, 3, 5-triazine scaffold is critical for drug potency. A comparison of the potency of compounds 6a and 7a-f revealed that the introduction of an additional ketone moiety reduced the antimicrobial activity. The introduction of some halogen groups on the phenyl ring generally increased the antimicrobial activity relative to that of the nonsubstituted derivatives. Notably, the position of the Cl substituent on the phenyl ring affected the potency, with activity in the order of 2, 4-(Cl)2 > 4-Cl ≥ 3-Cl. Isosteric replacement with a morpholine or dimethylamine group at the N-position of the dihydro-1, 3, 5-triazine ring generally did not affect the antimicrobial activity. It is noteworthy that compounds bearing a 2, 4-Cl2 substituted phenyl ring showed excellent antimicrobial activities. Therefore, these results provide further evidence that the 2, 4-Cl2 substituted benzene ring plays a critical role in the antimicrobial activity of these compounds, which is consistent with the results obtained for a previously reported series of rhodamine and aminoguanidine derivatives [12, 13].
|
|
Table 1 Inhibitory activity (MIC, mmol/L) of compounds 6a, 7a–f, 10a–b, 11a–c, 12a–c, 13a–b, 14a–c, 15a–c and 16a–c against various bacteria. |
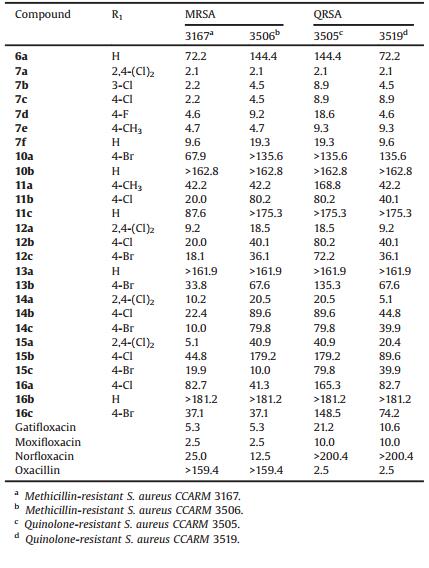
As indicated in Table 2, the inhibitory activities of all the synthesized compounds were tested against the clinical isolates of several different multidrug-resistant, Gram-positive bacterial strains (MRSA CCARM 3167 and 3506, and quinolone-resistant S. aureus (QRSA) CCARM 3505 and 3519). Compound 7a exhibited the most potent activity against the MRSA and QRSA strains, with an MIC of 2.1 mmol/L, making it equipotent or more potent than gatifloxacin and norfloxacin (MICs of 8 and 4 mg/mL against MRSA 3167 and 3506, respectively). In general, against multidrugresistant strains, series 7 compounds were much more potent than the compounds inthe otherseries. Compounds 7a, 7b, 7d, and 14a exhibited 2 to 4-fold greater activities, with MICs of 2.1-4.1 mmol/L, than gatifloxacin and moxifloxacin (MICs=10.0-21.2 mmol/L), and the activity of compound 7a was equal to that of oxacillin (MIC=2.5 mmol/L).
|
|
Table 2 Inhibitory activity (MIC, mmol/L) of compounds 6a, 7a–f, 10a–b, 11a–c, 12a–c, 13a–b, 14a–c, 15a–c and 16a–c against clinical isolates of multidrug-resistant Grampositive strains. |
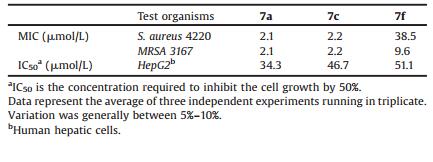
To see whether the antibacterial activity of the synthesized compounds were selectively toxic, we evaluated the cytotoxicity of compound 7a, 7c and 7f using a standard technique. As shown in Table 3, compound 7f exhibited weakeractivity than 7c against the different bacteria, in spite of its slight greater cytotoxicity than 7c, comparably indicating that the promising antibacterial activity of these compounds may not be due to their cytotoxicity, but some unknown mechanism of action.
|
|
Table 3 Antibacterial activity and cytotoxicity for 7a, 7c and 7f. |
{kind=link}
{kind=link}
To rationalize the observed antibacterial activity and understand the possible mechanism of action of these compounds, a libdocking investigation was undertaken. The crystal structure data (S. aureus DHFR) were obtained from the protein data bank (PDB ID: 3fra) [14, 15]. Enzyme structures were checked for missing atoms, bonds and contacts. Hydrogen atoms were added to the enzyme structure. Water molecules and bound ligands were manually deleted. Preferred coordination modes of 7a and 7f with dihydrofolate reductase (DHFR) protein are presented in Fig. 3. Fragment B of 7f is bound into the active site, in which the benzyl group shows H-bond interaction with Val 31 and Ala 7. Fragment A of 7a is bound into the active site where the morpholino group shows H-bond interaction with Phe 92, Leu 5, and Val 31, comparably indicated its more potent activity than that of 7f. The preliminary docking results imply that compounds 7a and 7f possibly display their antibacterial activity through the interaction with DHFR protein by targeting residues of the active cavities of DHFR.

|
Download:
|
| Fig. 3. Docking result of compound 7a and 7f. (A) Key residues in binding site surrounding 7a. (B) 2D molecular docking modeling of compound 7a with 3fra. (C) Key residues in binding site surrounding 7f. (D) 2D molecular docking modeling of compound 7f with 3fra. | |
{kind=link}
3. Conclusions
Based on our previous work, we synthesized a series of 1, 4-dihydro-1, 3, 5-triazine derivatives and evaluated their antibacterial activities against several Gram-positive and Gram-negative bacteria, as wellas a fungus (C. albicans). Some of these compounds exhibited high inhibitory activities against the Gram-positive and Gram-negative bacteria, including several multidrug-resistant clinical isolates. Compound 7a showed the most potent antimicrobial activity (MIC=2.1 mmol/L) against selected MRSA and QRSA strains. Preliminary docking study showed that these compounds have a good interaction with the active cavities of DHFR, possibly exhibit their potency via inhibiting DHFR. Further study examining the mechanisms of action of these compounds is currently underway in our laboratory.
4. ExperimentalThe synthesis of the target compounds is presented inScheme 1. A series of intermediates (4, 5, 8, and 9) was obtained by reacting hydroxybenzaldehyde or vanillin with the appropriate substituted benzyl chlorides or 2-bromo-1-phenylethanones [16]. Then, the intermediates were reacted with moroxydine hydrochloride or metformin hydrochloride in acetic acid to generate the target compounds. The structures of the synthesized compounds were confirmed by 1H NMR, 13C NMR, and mass spectroscopy (see Supporting information).

|
Download:
|
| Scheme 1. Synthetic scheme for the synthesis of compounds 6a, 7a–f, 10a–b, 11a–c, 12a–c, 13a–b, 14a–c, 15a–c and 16a–c. Reagents and conditions (a) K2CO3, DMF, 60 ℃ (b) AcOH, 120 ℃, 4–8 h. | |
{kind=link}
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81260468, 81460524 and 81060257) and the Natural Science Foundation of Jilin Province (No. 20160101218JC).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.05.022.
| [1] | J.N. Pendleton, S.P. Gorman, B.F. Gilmore. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 11 (2013) 297–308. DOI:10.1586/eri.13.12 |
| [2] | M.A. Fischbach, C.T. Walsh. Antibiotics for emerging pathogens. Science 325 (2009) 1089–1093. DOI:10.1126/science.1176667 |
| [3] | A. Khalafi, - Nezhad, M.N. Soltani Rad, H. Mohabatkar, Z. Asrari. B. Hemmateenejad. Bioorg. Med. Chem. 13 (2005) 1931–1938. DOI:10.1016/j.bmc.2005.01.014 |
| [4] | B.I. Schweitzer, A.P. Dicker, J.R. Bertino. Dihydrofolate reductase as a therapeutic target. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 4 (1990) 2441–2452. |
| [5] | J.M. Blaney, C. Silipo, A. Vittoria. Structure-activity relationships of dihydrofolate reductase inhibitors. Chem. Rev. 84 (1984) 333–407. DOI:10.1021/cr00062a002 |
| [6] | J.H. Feng, T. Ding, X.L. Ju. Synthesis and fungicidal activity of novel 1, 3, 5-triazine derivatives. J. Wuhan Inst. Tech. 35 (2013) 35–38. |
| [7] | B. Singha, H.R. Bhata, M.K. Kumawatb. Structure-guided discovery of 1, 3, 5-triazine-pyrazole conjugates as antibacterial and antibiofilm agent against pathogens causing human diseases with favorable metabolic fate. Bioorg. Med. Chem. Lett. 24 (2014) 3321–3325. DOI:10.1016/j.bmcl.2014.05.103 |
| [8] | Z.H. Chen, C.J. Zheng, L.P. Sun, H.R. Piao. Synthesis of new chalcone derivatives containing a rhodanine-3-acetic acid moiety with potential anti-bacterial activity. Eur. J. Med. Chem. 45 (2010) 5739–5743. DOI:10.1016/j.ejmech.2010.09.031 |
| [9] | M.X. Song, C.J. Zheng, X.Q. Deng, et al., Synthesis and bioactivity evaluation of rhodanine derivatives as potential anti-bacterial agents. Eur. J. Med. Chem. 54 (2012) 403–412. DOI:10.1016/j.ejmech.2012.05.023 |
| [10] | X. Jin, C.J. Zheng, M.X. Song, et al., Synthesis and antimicrobial evaluation of Lphenylalanine-derived C5-substituted rhodanine and chalcone derivatives containing thiobarbituric acid or 2-thioxo-4-thiazolidinone. Eur. J. Med. Chem. 56 (2012) 203–209. DOI:10.1016/j.ejmech.2012.08.026 |
| [11] | J. Miao, C.J. Zheng, L.P. Sun, et al., Synthesis and potential antibacterial activity of new rhodanine-3-acetic acid derivatives. Med. Chem. Res. 22 (2013) 4125–4132. DOI:10.1007/s00044-012-0417-z |
| [12] | M.X. Song, C.J. Zheng, X.Q. Deng, Z.Y. Wei, H.R. Piao. The synthesis and antibacterial activities of N-carboxymethyl rhodanines. Med. Chem. 4 (2014) 441–448. |
| [13] | Z.Y. Wei, K.Q. Chi, Z.K. Yu, et al., Synthesis and biological evaluation of chalcone derivatives containing aminoguanidine or acylhydrazone moieties. Bioorg. Med. Chem. Lett. 26 (2016) 5920–5925. DOI:10.1016/j.bmcl.2016.11.001 |
| [14] | C. Oefner, M. Bandera, A. Haldimann, et al., Increased hydrophobic interactions of iclaprim with Staphylococcus aureus dihydrofolate reductase are responsible for the increase in affinity and antibacterial activity. J. Antimicrob. Chemother. 63 (2009) 687–698. DOI:10.1093/jac/dkp024 |
| [15] | T. Lam, M.T. Hilgers, M.L. Cunningham, et al., Structure-Based design of new dihydrofolate reductase antibacterial agents:7-(benzimidazol-1-yl)-2, 4-diaminoquinazolines. J. Med. Chem. 57 (2014) 651–668. DOI:10.1021/jm401204g |
| [16] | S.J. Won, C.T. Liu, L.T. Tsao, et al., Synthetic chalcones as potential antiinflammatory and cancer chemopreventive agents. Eur. J. Med. Chem. 40 (2005) 103–112. DOI:10.1016/j.ejmech.2004.09.006 |