 2017, Vol. 28
2017, Vol. 28 



b Key Laboratory of Standard Material in Natural Medicine of Guangdong Province, Guangzhou Xiangxue Pharmaceutical Ltd. Co., Guangzhou 510663, China;
c Faculty of Chinese Medicine, Macau University of Science and Technology, Macao, China;
d State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Macao, China
Cynanchum stauntonii (Decne.) Schltr.exLévl., belonging to the family of Asclepiadaceae, is a common herb medicine, which has been used as antitussive and expectorant in China for a long time [1]. It is recorded as "Bai-Qian" in the Chinese Pharmacopoeia (ChP), together with the other species of the same genus, C. glaucescens (Decne.) Hand.-Mazz. Chemical investigations showed that they contained characteristic C21 steroidal glycosides, especially with the aberrant 13, 14:14, 15-disecopregnane-type skeleton or 14, 15-secopregnane-type skeleton, which exhibited anti-inflammatory, anti-virus and antitumor activities [2-6].
In our ongoing search for anti-inflammatory constituents from the roots of C. stauntonii [5-7], four new C21 steroidal glycosides (1-4), together with one known compound stauntoside F (5), were obtained from the 95% aqueous ethanol extract of the dried roots of C. stauntonii. Herein, we described the isolation and structural elucidation of new compounds (1-4). In addition, the antiinflammatory and cardiomyocyte protective effects of compounds 1-4 were evaluated.
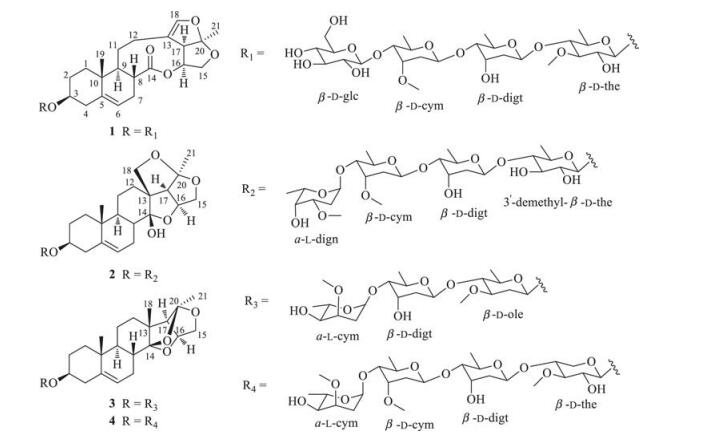
2. Results and discussionCompounds 1-4 were obtained as colorless amorphous powders (Fig. 1). They showed positive reactions to LibermannBurchard and Keller-Kiliani reagents, which suggested that they were likely steroidal glycosides with 2-deoxysugar substitutions [6, 7].

|
Download:
|
| Fig. 1. Chemical structures of steroidal glycosides 1–4. | |
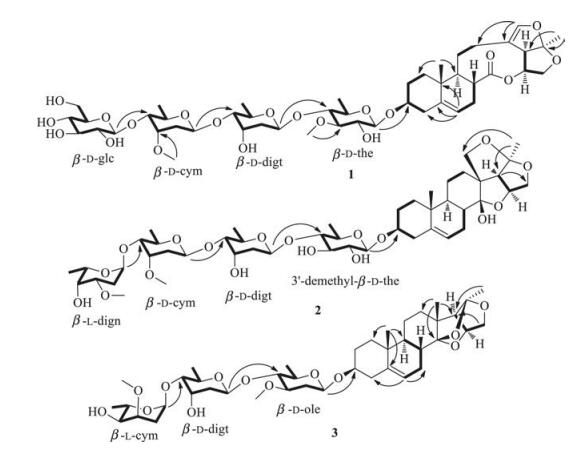
The HRESI-MS data of steroidal glycoside 1 showed the pseudomolecular ion at m/z 979.4545 [M + Na]+, suggesting that it had the molecular formula C47H72O20 (Calcd. for C47H72O20Na 979.4539) and accounted for 12 indices of hydrogen deficiency. The IR spectrum indicated the presence of hydroxy (3441 cm-1), carbonyl (1735 cm-1), and olefinic (1645 cm-1) functionalities. The 1H NMR spectrum (400 MHz, in C5D5N) showed two tertiary methyl groups at δH 0.78 (s, 3H, H-19) and 1.55 (s, 3H, H-21) and two olefinic protons at δH 5.33 (d, 1H, J = 5.0 Hz, H-6) and 6.48 (s, 1H, H-18). The 13C NMR spectrum showed an ester carbonyl signal at δC 175.8 (C-14), four olefinic carbon signals at δC 120.8 (C-6), 141.0 (C-5), 114.8 (C-13) and 144.2 (C-18), and two tertiary methyl signals at δC 18.2 (C-19) and 25.2 (C-21). Comparison of the 1H and 13C NMR data with that of stauntoside O [8] suggested that 1 had the aglycone of glaucogenin C, which was confirmed by 1D and 2D NMR data(Fig. 2). The 1H NMR spectrum of 1 showed four anomeric proton signals at δH 4.81(d, 1H, J=7.7Hz), 4.91 (d, 1H, J=7.8Hz), 5.11(d, 1H, J=9.3Hz), and 5.50 (d, 1H, J=9.1Hz), and three secondary methyl protonsignalsat δH1.38(d, 3H, J=6.0Hz), 1.46(d, 3H, J=5.4Hz), and 1.55 (d, 3H, J=6.0Hz), indicating that it had four sugar units, including three 6-deoxysugars. The 13C NMR spectrum displayed four anomeric carbon signals at δC 102.7 (C-1'), 99.3 (C-1''), 100.1 (C-1'''), and 106.9 (C-1'''') and three methyl carbon signals at δC 19.0 (C-6'), 18.9 (C-6''), and 18.8 (C-6'''), which confirmed that it was a tetraglycoside, including three 6-deoxysugars. The composition of sugar units was identified to be thevetose, digitoxose, cymarose, and glucose by comparison of its 1H and 13C NMR data with that of stauntosideL [9], which was confirmed by mild acid hydrolysisand subsequent silica gel column chromatography purification. The D-configuration of glucose was determined by acid hydrolysis and appropriate derivatization of the resulting sugar [10]. The d-configurations of cymarose, thevetose, and digitoxose were determined by comparison of their specific rotation with that of literatures[5, 11-15] after24h equilibrationin anaqueous solution. The β orientation of all the sugar units was identified by their large J values of 3JH-1', H-2' (J > 7 Hz). The HMBC correlations (Fig. 2) from δH 4.91 (H-1'''' of glucose) to δC 83.2 (C-4''' of cymarose), δH 5.11 (H-1''' of cymarose) to δC 83.4 (C-4'' of digitoxose), and δH 5.50 (H-4'' of digitoxose) to δC 83.2 (C-4' of thevetose) suggested that the sugar units were 1→4 linkages. The HMBC correlation (Fig. 2) between δH 4.81 (d, 1H, J = 7.7 Hz, H-10 of thevetose) and δC 78.7 (C-3) indicated that the sugar chain was attached to C-3 position of glaucogenin C. Thus, steroidal glycoside 1 was identified tobe glaucogeninC 3-O-β-D-glucopyranosyl-(1→4)-β-D-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-D-thevetopyranoside. Full assignment of its 1H and 13C NMR data was achieved by analyses of 1H, 13C NMR, 1H-1H COSY, HSQC, and HMBC spectra (Table 1).

|
Download:
|
| Fig. 2. Key 1H-1H COSY and HMBC correlations of steroidal glycosides 1–3. | |
|
|
Table 1 1H NMR and 13C NMR data for steroidal glycosides 1–4 (J values in Hz). |

{kind=link}
{kind=link}
Steroidal glycoside 2 had the molecular formula C47H74O18 by its pseudomolecular ion at m/z 949.4760 [M + Na]+ (Calcd. for C47H74O18Na 949.4773) in its HRESI-MS and accounted for 11 indices of hydrogen deficiency. The IR spectrum showed the absorption bands for hydroxy (3457 cm-1) and olefinic (1646 cm-1) functionalities. The 1H NMR spectrum (300 MHz in CDCl3) displayed two tertiary methyl signals at δH 0.91 (s, 3H, H-19) and 1.44 (s, 3H, H-21), one olefinic proton signal at δH 5.39 (d, 1H, J = 4.9 Hz, H-6), two oxygen-substituted methine proton signals at δH 3.49 (m, 1H, H-3) and 4.69 (q, 1H, J = 3.7 Hz, H-16), and two oxygen-substituted methylene proton signals at δH 4.16 (d, 1H, J = 10.9 Hz, H-15a)/3.72 (t, 1H, J = 4.0 Hz, H-15b) and 4.48 (d, 1H, J = 9.0 Hz, H-18a)/3.75 (m, 1H, H-18b). The 13C NMR spectrum showed two olefinic carbon signals at δC 122.6 (C-6) and 139.3 (C-5), two oxygenated methine carbon signals at δC 79.0 (C-3) and 80.7 (C-16), two oxygenated methylene carbon signals at δC 75.3 (C-15) and 77.9 (C-18), and two tertiary methyl carbon signals at δC 18.3 (C-19) and 22.5 (C-21). Comparison of its 1H and 13C NMR data with that of hirundigosides E-J [7] suggested that the aglycone of steroidal glycoside 2 was hirundigenin, which was confirmed by the presence of hemiketal carbon signal at δC 108.3 (C-14). The anomeric proton signals at δH 4.33 (d, 1H, J = 7.8 Hz), 4.75 (dd, 1H, J = 7.5, 1.5 Hz), 4.78 (dd, 1H, J = 7.4, 1.5 Hz), and 4.95 (d, 1H, J = 3.2 Hz) suggested that 2 was a tetraglycoside in the 1H NMR spectrum, which was confirmed by the anomeric carbon signals at δC 100.8 (C-1'), 99.3 (C-1''), 98.3 (C-1'''), and 100.7 (C-1'''') in the 13C NMR spectrum. The four sugar units were 6-deoxysugars by the presence of four secondary methyl signals at δH 1.23 (d, 3H, J = 5.8 Hz, H-60)/δC 17.8 (C-6'), 1.21 (d, 3H, J = 5.6 Hz, H-600)/18.0 (C-600), 1.16 (d, 3H, J = 6.3 Hz, H-6000)/18.4 (C-6'''), and 1.26 (d, 3H, J = 6.8 Hz, H-6'''')/17.2 (C-6'''') in its 1H and 13C NMR spectra. The composition of sugar units was identified to be 3-demethylthevetose, digitoxose, cymarose, and diginose by comparison of its 1H and 13C NMR data with that of stauntoside A and hirundigoside G [7, 16]. Mild acid hydrolysis of 2 and subsequent silica gel column chromatography purification led to obtain the monosugars, which confirmed the deduction mentioned above. The l-configuration of diginose and d-configurations of 3-demethyl-thevetose, digitoxose, and cymarose were identified by comparing their specific rotation with literatures [5, 11, 13-15]. The orientation of anomeric proton for diginose was identified to be α by the small value of 3JH-1', H-2' (J = 3.2 Hz). The β orientations of 3-demethyl-thevetose, digitoxose, and cymarose were determined by their large splitting values of 3JH-1', H-2' (J = 7.5, 7.8, and 9.5 Hz, respectively). The 1→4 linkages of sugar units were identified by the HMBC correlations (Fig. 2) from δH 4.95 (H-10000 of diginose) to δC 81.7 (C-4'''), from δH 4.75 (H-1''' of cymarose) to δC 82.0 (C-4''), and from δH 4.78 (H-100 of digitoxose) to δC 87.0 (C-40 of 3-demethyl-thevetose). The HMBC correlation (Fig. 2) between δH 4.33 (H-1' of 3-demethylthevetose) and δC 79.0 (C-3) attached the sugar chain to the C-3 position of aglycone. Thus, steroidal glycoside 2 was identified to be hirundigenin 3-O-α-L-diginopyranosyl-(1→4)-β-D-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-d-3'- demethylthevetopyranoside, and named Hirundigoside K. Full assignment of 1H and 13C NMR data was achieved by analyses of 1H, 13C NMR, 1H-1H COSY, HSQC, and HMBC spectra (Table 1).
Steroidal glycoside 3 had a molecular formula C41H64O13 by the pseudomolecular ion at m/z 787.4249 [M + Na]+ (Calcd. for C41H64O13Na 787.4245) and accounted for 10 indices of hydrogen deficiency. The IR spectrum showed the absorption bands for hydroxy (3442 cm-1) and methyl (2939 cm-1) functionalities. The 1H NMR spectrum (300 MHz, in C5D5N) showed the presence of one olefinic proton at δH 5.44 (m, 1H, H-6) and three tertiary methyl protons at δH 1.00 (s, 3H, H-19), 1.11 (s, 3H, H-18), and 1.75 (s, 3H, H-21). The 13C NMR spectrum showed the presence of two ketal carbon signals at δC 109.5 (C-14) and 114.1 (C-20) and three methyl carbon signals at δC 16.0 (C-18), 19.7 (C-19), and 24.4 (C-21). Comparison of the 1H and 13C NMR data with that of stauntoside T [5] suggested that they had the same aglycone, which was confirmed by key 1H-1H COSY and HMBC correlations (Fig. 2). The presence of three anomeric proton signals at δH 4.83 (dd, 1H, J = 9.7, 1.8 Hz), 5.10 (dd, 1H, J = 4.1, 2.1 Hz), and 5.46 (dd, 1H, J = 9.7, 1.8 Hz) in the 1H NMR spectrum suggested that it was a triglycoside, which was confirmed by the presence of three anomeric carbon signals at δC 98.5 (C-1'), 98.9 (C-1'''), and 99.0 (C-1'') in the 13C NMR spectrum. Steroidal glycoside 3 had the same sugar chain as that of hirundigoside G [7] by comparison of their 13C NMR data. Mild acid hydrolysis of 3 and subsequent silica gel chromatography purification led to obtain cymarose, digitoxose, and oleandrose [5]. The l-configuration of cymarose and d-configurations of digitoxose and oleandrose were determined by comparing their specific rotation with that of previous reports [5, 13-15]. The a orientation of l-cymarose and β orientations of d-digitoxose and doleandrose were identified by their splitting values of 3JH-1', H-2' (J = 4.1, 9.7, and 9.7 Hz, respectively). In the HMBC spectrum, correlations (Fig. 2) from δH 5.10 (H-1''' of l-cymarose) to δC 81.3 (C-4'') and from δH 5.46 (H-1'' of d-digitoxose) to δC 83.6 (C-4') revealed that the sugar chain was 1→4 linkages. The HMBC correlation (Fig. 2) between δH 4.83 (H-1' of d-oleandrose) and δC 78.0 (C-3) attached the sugar chain to C-3 position of the aglycone. The daughter ions at m/z 621.3644 [M-cym + H]+, 491.3013 [Mcym-digt + H]+, and 347.2227 [M-cym-digt-ole + H]+ in the HR ESIMS/MS confirmed the deduction mentioned above. Thus, steroidal glycoside 3 was identified to be (14S, 16S, 20R)-14, 16-14, 20-15, 20-triepoxy-14, 15-secopregn-5-en-3-ol-3-O-α-L-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-D-oleandropyranoside, and named stauntoside U. Full assignment of 1H and 13C NMR data was achieved by analyses of 1H and 13C NMR, HSQC, 1H-1H COSY, and HMBC spectra (Table 1).
Steroidal glycoside 4 had the molecular formula C48H76O17 by the pseudomolecular ion at m/z 947.4972 [M+Na]+ (Calcd. for C48H76O17Na 947.4980) and accounted for 11 indices of hydrogen deficiency. The 1H NMR spectrum (300 MHz, in C5D5N) of 4 showed the presence of one olefinic proton at δH 5.35 (m, 1H, H-6) and three tertiary methyl protons at δH 0.93 (s, 3H, H-19), 1.10 (s, 3H, H-18), and 1.74 (s, 3H, H-21). Comparison of the 1H and 13C NMR data with that of steroidal glycoside 3 suggested that they had the same aglycone and the difference lay in the composition of the sugar chain, which was confirmed by the 1H-1H COSY and HMBC correlations. The presence of four anomeric proton signals at δH 4.85 (d, 1H, J = 7.7 Hz), 5.16 (dd, 1H, J = 9.2, 1.0 Hz), 5.22 (d, 1H, J = 3.7 Hz), and 5.54 (dd, 1H, J = 9.7, 1.8 Hz) showed that it was a tetraglycoside, which was confirmed by the presence of anomeric carbon signals at δC 99.4 (C-1''), 100.1 (C-1'''), 101.7 (C-1''''), and 102.7 (C-1'). Full assignment of the 1H and 13C NMR data was achieved by analyses of 1H, 13C NMR, HSQC, 1H-1H COSY, and HMBC spectra (Table 1). Steroidal glycoside 4 had the same sugar chain as that of hirundigoside J [7] by comparison of their 1H and 13C NMR data. Mild acid hydrolysis and subsequent silica gel chromatography purification led to obtain diginose, cymarose, digitoxose, and thevetose [5]. The absolute configuration of cymarose was identified to be L, while the other three sugars were identified to be D configurations by their specific rotations [5, 11-15]. The α orientation of L-diginopyranose and β orientations of the other three sugars were determined by their splitting values of 3JH-1', H-2'. The 1→4 linkage of sugar chain was identified by the HMBC correlations from δH 5.22 (H-1'''') to δC 82.7 (C-4'''), from δH 5.16 (H-1''') to δC 83.7 (C-4''), and from δH 5.54 (H-1'') to δC 83.4 (C-4'). The HMBC correlation between δH 4.85 (H-1') and δC 78.7 (C-3) attached the sugar chain to C-3 position of the aglycone. Thus, steroidal glycoside 4 was identified to be (14S, 16S, 20R)-14, 16-14, 20-15, 20-triepoxy-14, 15-secopregn-5-en-3-ol-3-O-α-L-cymaropyranosyl-(1→4)-β-D-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-D-thevetopyranoside, and named stauntoside V.
The known steroidal glycoside 5 was identified to be stauntoside F by comparing its physical and spectroscopic data with that reported previously [17].
The anti-inflammatory effect of new compounds 1-4 toward LPS-induced RAW264.7 macrophages was investigated since our previous studies showed that C21 steroidal glycosides from Cynanchum stauntonii exhibited moderate anti-inflammatory activity [5-7]. We found that they could not inhibit LPS-induced iNOS and COX-2 protein expression compared to LPS stimulation alonein RAW264.7 cellsat aconcentrationof 30 mmol/L, indicating that they did not have the potential to mediate anti-inflammatory effects (data not shown). On the contrary, compounds 1-4 could increase expression of COX-2 in RAW264.7 macrophages, suggesting that they may display the potential of cardiomyocyte protection [18-20]. Next, cytotoxicity of compounds 1-4 was evaluated by measuring the cell viability by the MVS method. The H9c2 cells were pretreated with different compounds (10 mmol/L) for 24h, cytotoxicity evaluation showed that no compounds exhibited cytotoxicity (Fig. 3). We then evaluated their protective effects toward H9c2 cells injury induced by hypoxia/reoxygenation (H/R) model. Diazoxide was used as positive control. Results revealed that compounds 1-4 have significant protective effects against H/R injury compared to Diazoxide, while compound 4 showed a dose dependent manner in treating H/R induced cell death (Fig. 4). Based on these results, we concluded that compounds 1-4 showed significant cardioprotective effects and their underlying mechanisms of action deserves further study.

|
Download:
|
| Fig. 3. The cytotoxic effect of compounds 1-4 in H9c2 cells under normoxic conditions. The normal group was always maintained in normoxic condition. All measurements were repeated in three wells, the results are the average of four independent experiments. All values are expressed as mean ± SD. | |
{kind=link}

|
Download:
|
| Fig. 4. Protective effects of compounds 1-4 against H/R injury in H9c2 cells. The normal group was always maintained in normoxic condition. All measurements were repeated in three wells, the results are the average of four independent experiments. All values are expressed as mean ± SD. N = 3. ^^^ P < 0.001 vs. Normal, *** P < 0.001 vs. H/R. | |
{kind=link}
3. Conclusion
Four new C21 steroidal glycosides 1-4 were obtained from the dried roots of C. stauntonii. Among them, compound 1 was interesting 13, 14:14, 15-disecopregnane-type skeleton C21 steroidal glycosides that contained glaucogenin C aglycone; while compounds 2-4 were interesting 14, 15-secopregnane-type skeleton C21 steroidal glycosides, in which was hirundigenin type C21 steroidal glycoside. In our previous report, we found that this class of steroidal glycoside existed in nature as epimers owing to the presence of 14-hemiketal hydroxyl group in its structure [7]. Meanwhile, we also found that it was more striking that they always displayed two sets of 1H and 13C NMR signals when measured in pyridine-d5 while exhibiting one set of 1H and 13C NMR signals in CDCl3 with 14β-OH epimer. It may ascribe to the more favorable formation of intramolecular hydrogen bond due to the proximate distance of hydroxyl group and oxygen atom for 14β-OH epimer than that of 14α-OH epimer in CHCl3. In addition, pyridine, as a Lewis base, promotes the isomerization of hemiketal hydroxyl group of hirundigosides [7]. Although new compounds 1-4 did not exhibit anti-inflammatory effects on LPS stimulated RAW264.7 cells, they have significant protective effects toward H9c2 cells injury induced by hypoxia/reoxygenation (H/R). This showed that compounds 1-4 showed significant cardioprotective effects and their underlyingmechanisms of action deservesfurther study.
4. Experimental 4.1. General experimentalOptical rotations were obtained on a P-1020 digital polarimeter (Jasco Corporation, Tokyo, Japan). IR spectra were recorded on a Jasco FTIR-480 Plus spectrometer. NMR spectra were measured with Bruker AV 300, 400, and 600 NMR instruments. Chemical shifts were given in ppm (δ) relative to chemical shifts of solvent resonances(C5D5N: 8.74 and 150.3ppm, CDCl3: 7.24 and 77.2ppm). HRESI-MS was obtained with a Micromass Q-TOF mass spectrometer (Waters Corporation, Massachusetts, United States). HPLC was performed with a Sunfire C18 column (4.6mm ×250mm, 5 mm) and a HPLC system equipped with a Waterse2695 system, a Waters 2998 PDA detector and an Alltech3300 ELSD detector. Semipreparative HPLC was performed on a HPLC system equipped with a Waters 1515 pump and a Waters 2489 detector, using a Sunfire C18 column (10.0mm ×250mm, 5 mm). Open column chromatography (CC) was performed using silica gel (200-300mesh, Qingdao Haiyang Chemical Group Corp., Qingdao), ODS column (50 mm, YMC Co., LTD, Kyoto, Japan), and Sephadex LH-20 (Pharmacia Corp., United States). TLC analysis was performed on pre-coated silica gel GF254 plates (Qingdao Haiyang Chemical Group Corp., Qingdao).
4.2. Plant materialThe Chinese Medicine "Bai-Qian, " the dried roots of C.stauntonii, was obtained from Guangzhou Xiangxue Pharmaceutical Co., Ltd., in November, 2012. It was identified by associate Prof. Ying Zhang, College of Pharmacy, Jinan University, Guangzhou. A voucher specimen (CS201211-1) was deposited intothe Institute of Traditional Chinese Medicine and Natural Products, Jinan University.
4.3. Extraction and isolationThe dried and chopped roots (9.5kg) of C. stauntonii were refluxed three times in 95% (v/v) aqueous EtOH (90L ×3). The combined ethanol extract was evaporated under vacuum to yield a dark-brown residue (ca. 1100 g). The residue was directly chromatographed over an open macroporous Diaion HP 20 column (φ 12.0 cm × 63.0 cm) and eluted with a gradient of EtOH:H2O (0, 30, 50, and 95% of EtOH) to yield 4 fractions (A-D). Fraction D (93.0 g, 95% ethanol) was applied to a silica gel column chromatography (φ 9.5 cm × 68.0 cm) using a gradient of mixtures of petroleum ether:EtOAc (100:0, 98:2, 95:5, 9:1, 7:3, 0:100) and EtOAc:methanol (CH3OH) (95:5, 9:1, 7:3, 0:100) to yield 13 subfractions (D1-D13) according to their TLC profiles and HPLC behavior. Fraction D10 (20.8 g) was subjected to open ODS CC (φ 4.0 cm × 30.0 cm) using a CH3OH:H2O gradient to afford 13 subfractions (D10-1-D10-13). Subfraction D10-6 was subjected to ODS CC using a gradient of CH3OH:H2O as the eluent to yield 8 subfractions (D10-6-1-D10-6-8). Subfraction D10-6-7 was subjected to Sephadex LH-20 CC (CH3OH) and then applied to a semipreparative RP-HPLC system (CH3OH:H2O 70:30) to afford compound 4 (10.0 mg). Subfraction D10-7 was separated by ODS CC using gradient solvents of CH3OH:H2O to yield 7 subfractions (D10-7-1-D10-7-7). Subfraction D10-7-6 was subjected to Sephadex LH-20 CC (CH3OH) and then applied to a semi-preparative RPHPLC system (CH3OH:H2O 70:30) to yield compound 3 (8.0 mg). Subfraction D12 (24.0 g) was subjected to ODS CC (φ 4.0 cm × 30.0 cm) using a gradient of CH3OH:H2O to afford 13 subfractions (D12-1~D12-13). Subfraction D12-2 was applied to Sephadex LH-20 CC (CH3OH:H2O 70:30) and then subjected to a semi-preparative RPHPLC system (CH3OH:H2O 65:35) to obtain compound 2 (35.0 mg). Subfraction D12-3 was subjected to silica gel CC (φ 2.5 cm × 25.0 cm) eluted with CHCl3:CH3OH (95:5, 90:10, 85:15, and 80:20) to yield seven subfractions (D12-3-1~D12-3-7). Subfraction D12-3-6 was subjected to a semi-preparative RP-HPLC system (CH3OH:H2O 70:30) to yield compound 1 (51.0 mg).
4.4. Mild acid hydrolysis of compounds 1-4 and determination of the absolute configuration of monosugarsMild acid hydrolysis and subsequent silica gel CC of compounds 1-4 were achieved following previous methods [5]. Each compound (1-4, 5-20 mg) in CH3OH (2 mL) was mixed with 0.05 mol/L HCl (3 mL), stirred for 15 min at 70 ℃ and then evaporated in vacuum to remove CH3OH. The aqueous solution was extracted with CHCl3 (3 × 3 mL). The aqueous layer was neutralized with Ba(OH)2 saturated with H2O. The precipitates were filtered out and the solution was dried to yield a crude sugar fraction. The crude sugar fraction was subjected to silica gel CC, successively eluting with CHCl3:CH3OH to yield thevetose (85:15, v/v), digitoxose (93:7, v/v), cymarose (97:3, v/v), 3-demethylthevetose (80:20, v/v), diginose (98:2, v/v), oleandrose (97:3, v/v) and glucose (0:100).
Glaucogenin C 3-O-β-D-glucopyranosyl-(1→4)-β-D-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-D-thevetopyranoside (1): colorless amorphous powder; [α]D27 -29.7 (c 0.5, CH3OH); IR (KBr) νmax 3441, 2933, 1735, 1645, and 1060 cm-1; ESI-MS (positive mode) m/z 979.9 [M + Na]+, HRESI-MS (positive mode) m/z 979.4545 [M + Na]+ (calcd. for C47H72O20Na, 979.4539); 1H (400 MHz, C5D5N) and 13C NMR (100 MHz, C5D5N) data, see Table 1.
Hirundigenin 3-O-α-L-diginopyranosyl-(1→4)-β-D-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-D-30-demethylthevetopyranoside (Hirundigoside K, 2): colorless amorphous powder; [α]D27 -50.6 (c 0.5, CH3OH); IR (KBr) νmax3457, 2973, 1646, and 1062 cm-1; ESI-MS (positive mode) m/z 949.9 [M + Na]+, HRESI-MS (positive mode) m/z 949.4760 [M + Na]+ (calcd. for C47H74O18Na, 949.4773); 1H (300 MHz, CDCl3) and 13C NMR (75 MHz, CDCl3) data, see Table 1.
(14S, 16S, 20R)-14, 16-14, 20-15, 20-Triepoxy-14, 15-secopregn-5-en-3-ol-3-O-α-L-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-D-oleandropyranoside (stauntoside U, 3): colorless amorphous powder, [α]D27 -66.7 (c 0.5, CH3OH). IR (KBr) νmax: 3442, 2939, 1538, 1457, 1063 cm-1. ESI-MS (positive mode) m/z 787.6 [M + Na]+. HRESI-MS (positive mode) m/z 787.4249 [M + Na]+ (calcd. for C41H64O13Na 787.4245). 1H (300 MHz, C5D5N) and 13C NMR (75 MHz, C5D5N) data: see Table 1.
(14S, 16S, 20R)-14, 16-14, 20-15, 20-Triepoxy-14, 15-secopregn-5-en-3-ol-3-O-α-L-diginopyranosyl-(1→4)-β-D-cymaropyranosyl-(1→4)-β-D-digitoxopyranosyl-(1→4)-β-D-thevetopyranoside (stauntoside V, 4): colorless amorphous powder, [α]D27 -28.8 (c 0.5, CH3OH). IR (KBr) νmax: 3506, 2936, 1548, 1455, 1062 cm-1. ESI-MS (positive mode) m/z 947.7 [M + Na]+. HRESI-MS (positive mode) m/z 947.4972 [M + Na]+ (calcd. for C48H76O17Na 947.4980). 1H NMR (300 MHz, C5D5N) and 13C NMR (75 MHz, C5D5N) data: see Table 1.
4.5. Biological studies 4.5.1. Cell cultureThe rat ventricular myocardial cell line H9c2 was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 mg/mL streptomycin at 37 ℃ under normoxic condition (95% air/5% CO2).
4.5.2. MVS assay for cell viabilityThe cell viability was measured by MVS assay. In brief, after cell culture was finished, each of the H9c2 cells with 100 mL of medium was overlaid with 2 × mitochondrial viability stain (100 mL), and then the cells were put in the incubator with the normoxic environment (95% air/5% CO2) for 4 h in the dark. The fluorescent intensity was measured at excitation 550 nm and emission 590 nm by a TECAN Infinite M200 micro plate reader (Tecan, Durham, USA).
4.5.3. Hypoxia/reoxygenation(H/R) model and sample treatmentTo examine the effectiveness of compounds 1-4 on H9c2 cells under hypoxia/reoxygenation condition, the cells were cultured under normoxic condition (95% air/5% CO2) overnight; then the cells were washed twice with Krebs-Ringer Bicarbonate buffer (composition in mmol/L: NaCl 115, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 24, HEPES 10; pH 7.4) balanced with N2 at 4 ℃ for overnight. Aliquots (100 mL) of KRB supplemented with 0.01% (w/v) BSA were added to the cells immediately prior to hypoxia. To create a hypoxia condition, a hypoxic chamber (Stem Cell Technologies, USA) was used to produce an in vitro hypoxia challenge. In essence, the cells were placed in the sealed chamber and the chamber was flushed with pure N2 for 5 min at a flow rate of 20 mL/min, then, all sealable connectors were closed. After that, the chamber was transferred to an incubator and the cells in the chamber were subjected to 3 h incubation at 37 ℃ to induced hypoxia. Reoxygenation was initiated by opening the chamber and then replacing the KRB with fresh DMEM medium, the cells were then cultured in an incubator under normoxic condition (95% air/ 5% CO2) at 37 ℃ for another 4 h. Then, the cells were seeded at the density of 5 ×104 cells/well in a 96-well plate. Samples (final concentrations: 1, 3, 10 mmol/L, respectively) were added to the culture system 1 h before hypoxia treatment and throughout the reoxygenation period. DZ was use as positive control. The control group was always maintained in normal DMEM and put in the incubator under normoxic atmosphere (95% air/5% CO2).
4.5.4. Statistical evaluationAll data were given as the mean ± SD of mean for the number of experiments. Statistical significance was compared each treated with compounds group with the blank control and determined by using one-way ANOVA with Graph Pad Prism 6.0 software (Graph Pad Software Inc, La Jolla, CA, USA). P values less than 0.05 was accepted as statistically significant.
AcknowledgmentsWe are grateful to Ms. L. Wang, Ms. X.Y. Shen, Mr. D.D. Wang and Dr. Y. Yang for the HR ESI-MS and NMR measurements. Part of the chemical work was accomplished in Guangzhou Xiangxue Pharmaceutical Ltd., Co. This work was supported by grants from Science and Technology Planning Project of Guangdong Province, China (No. 2015B030301005).
| [1] | The State Pharmacopoeia Commission of PRC, Pharmacopoeia of the People's Republic of China, China Medical Science and Technology Press, Beijing, China, 2010, pp. 101. |
| [2] | M. Zhang, J.S. Wang, J. Luo, D.D. Wei, L.Y. Kong. Glaucogenin E, a new C21 steroid from Cynanchum stauntonii. Nat. Prod. Res. 27 (2013) 176–180. DOI:10.1080/14786419.2012.665914 |
| [3] | M. Shibano, A. Misaka, K. Sugiyama, M. Taniguchi, K. Baba. Two secopregnanetype steroidal glycosides from Cynanchum stauntonii (Decne.) Schltr.ex Levl. Phytochem. Lett. 5 (2012) 304–308. |
| [4] | Y.M. Li, L.H. Wang, S.L. Li, et al., Seco-pregnane steroids target the subgenomic RNA of alphavirus-like RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 8083–8088. DOI:10.1073/pnas.0702398104 |
| [5] | C.Z. Lai, J.X. Liu, S.W. Pang, et al., Steroidal glycosides from the roots of Cynanchum stauntonii and their effects on the expression of iNOS and COX-2. Phytochem. Lett. 16 (2016) 38–46. DOI:10.1016/j.phytol.2016.02.016 |
| [6] | J.X. Liu, J.S. Tang, Y.H. Zuo, et al., Stauntoside B inhibits macrophage activation by inhibiting NF-κB and ERK MAPK signaling. Pharmacol. Res. 111 (2016) 303–315. DOI:10.1016/j.phrs.2016.06.022 |
| [7] | C.Z. Lai, H.B. Liu, J.X. Liu, et al., Hirundigenin type C21 steroidal glycosides from Cynanchum stauntonii and their anti-inflammatory activity. RSC Adv. 6 (2016) 59257–59268. DOI:10.1039/C6RA11957C |
| [8] | T. Nakagawa, K. Hayashi, H. Mitsuhashi. Studies on the constituents of Asclepiadaceae plants. LⅢ. The structures of glaucogenin-A -B, and -C monoD-thevetoside from the Chinese drug "Pai-ch'ien", Cynanchumg laucescens Hand-Mazz. Chem. Pharm. Bull. 31 (1983) 870–878. DOI:10.1248/cpb.31.870 |
| [9] | J. Q. Yu, Z. H. Zhang, A. J. Deng, H. L. Qin, Three new steroidal glycosides from the roots of Cynanchum stauntonii, BioMed. Res. Int. (2013) Article ID 816145, 7 pages. |
| [10] | T. Tanaka, T. Nakashima, T. Ueda, K. Tomii, I. Kouno. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 55 (2007) 899–901. DOI:10.1248/cpb.55.899 |
| [11] | Y.B. Liu, W.Z. Tang, S.S. Yu, et al., Eight new C-21 steroidal glycosides from Dregeasinensis var. corrugate. Steroids 72 (2007) 514–523. |
| [12] | J.L. Li, J. Zhou, Z.H. Chen, et al., Bioactive C21steroidal glycosides from the roots of Cynanchum otophyllum that suppress the seizure-like locomotor activity of zebrafish caused by pentylenetetrazole. J. Nat. Prod. 78 (2015) 1548–1555. DOI:10.1021/np501058b |
| [13] | L. Wang, Z.Q. Yin, Y. Wang, et al., Perisesaccharides A-E, new oligosaccharides from the root barks of Periplocasepium. Planta Med. 76 (2010) 909–915. DOI:10.1055/s-0029-1240842 |
| [14] | H. Hidekazu, A. Hideaki, K. Keisuke, H. Akita. Nucleophilic addition to 2, 3-disubstituted butanal derivatives and their application to natural product synthesis. Chem. Pharm. Bull. 58 (2010) 1411–1418. DOI:10.1248/cpb.58.1411 |
| [15] | M. Brasholz, H.U. Reißig. Alkoxyallene-based de novo synthesis of rare deoxy sugars:new routes to L-cymarose, L-sarmentose, L-diginose and L-oleandrose. Eur. J. Org. Chem. 21 (2009) 3595–3604. |
| [16] | N.Q. Zhu, M.F. Wang, H. Kikuzaki, N. Nakatani, C.T. Ho. Two C21-steroidal glycosides isolated from Cynanchum stauntonii. Phytochemistry 52 (1999) 1351–1355. DOI:10.1016/S0031-9422(99)00439-2 |
| [17] | J.Q. Yu, A.J. Deng, H.L. Qin. Nine new steroidal glycosides from the roots of Cynanchum stauntonii. Steroids 78 (2013) 79–90. DOI:10.1016/j.steroids.2012.10.007 |
| [18] | H. Sato, R. Bolli, G.D. Rokosh, et al., The cardioprotection of the late phase of ischemic preconditioning is enhanced by postconditioning via a COX-2-mediated mechanism in conscious rats. Am. J. Physiol. Heart Circ. Physiol. 293 (2007) H2557–2564. |
| [19] | X. Jiang, E. Shi, Y. Nakajima, et al., Cyclooxygenase-1 mediates the final stage of morphine-induced delayed cardioprotection in concert with cyclooxygenase-2. J. Am. Coll. Cardiol. 45 (2005) 1707–1715. DOI:10.1016/j.jacc.2005.02.046 |
| [20] | H.J. Kwak, K.M. Park, H.E. Choi, H.Y. Park. Protective mechanisms of NO preconditioning against NO-induced apoptosis in H9c2 cells:role pf PKC and COX-2. Free Radic. Res. 43 (2009) 744–752. DOI:10.1080/10715760903040602 |