 2017, Vol. 28
2017, Vol. 28 



b State Key Laboratory of Drug Research, and Natural Products Chemistry Department, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zhangjiang Hi-Tech Park, Shanghai 201203, China
Caesalpinia sappan L. (Fabaceae) is widely distributed in tropical and subtropical regions, including Southeast Asia (Burma, Vietnam and Indonesia) and Southwest China [1]. The dried heartwood of the plant, named Sappan Lignum, has been used as anti-malarial, anti-inflammatory and analgesic agent in traditional Chinese medicine [2, 3]. The seeds of this plant are rich in cassane-type diterpenoids, characterized with tricyclic framework with fused furan or butenolide ring [4-6]. These diterpenoids were reported with wide bioactivities, including cytotoxicity, anti-inflammation and anti-malaria [7]. Our previous study led to the isolation of a series of cassane-type diterpenoids from the seeds of C. sappan [8-10]. As a part of our ongoing investigation on the discovery of new anti-cancer agents, five new diterpenoids (Fig. 1) were isolated from the seeds of C. sappan. Herein, the isolation and structure elucidation of the new compounds, as well as their cytotoxic effect on human breast cancer MCF-7 and human colon cancer HCT116 cell lines, were described.

|
Download:
|
| Fig. 1. Chemical structures of compounds 1–5 and 5a. | |
2. Results and discussion
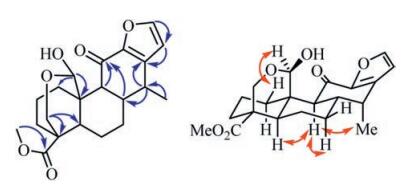
Compound 1, a white amorphous powder, had a molecular formula of C21H26O6 as determined by the protonated ion peak in the HR-ESIMS (m/z 397.1634 for C21H26O6Na, calcd. m/z 397.1627, see Supporting information Fig. S7), indicating nine degrees of unsaturation. The 1H and 13C NMR spectra of 1 displayed resonances for a 1, 2-disubstituted furan ring [δH 7.50 (d, 1H, J = 1.6 Hz) and 6.36 (d, 1H, J = 1.6 Hz); δC 147.3, 146.8, 143.8 and 110.5], an oxygenated methylene [δH 4.35 (dd, 1H, J = 11.7, 2.3 Hz) and 3.73 (d, 1H, J = 11.9 Hz); δC 61.3], an oxygenated methine [δH 5.72 (s, 1H); δC 95.3], a methoxyl [δH 3.68 (s, 3H); δC 51.6], a secondary methyl [δH 1.14 (d, 3H, J = 7.2 Hz); δC 13.9], and two carbonyl carbons (δC 187.2 and 175.6) (Table 1). These data revealed that 1 possessed a cassane-type furanoditerpene framework [11], and its structure was closely related to that of phanginin A [12]. The major difference was a methylene [δH 2.60 (dd, J = 15.9, 11.7 Hz), 2.72 (dd, J = 15.9, 5.7 Hz); δC 22.5] in phanginin A replaced by a carbonyl carbon (δC 187.2) in 1. The HMBC correlations of this carbonyl carbon with H-9 (δH 2.26) and H-8 (δH 3.08) assigned it as C-11 (Fig. 2). The relative configuration of 1 was inferred by ROESY spectrum (Fig. 2). The correlations of H-5/H-9, H-9/H-7a and H-9/ H3-17 indicated these protons and H3-17 were axial and α-oriented. The hydroxy group at C-20 was determined to be β-oriented based on the NOE correlations between H-20 (δH 5.72) and H-1β (δH 3.01) (Fig. 2). Accordingly, the structure of compound 1 was established, and it was named 11-oxophanginin A.
|
|
Table 1 1H (600 MHz, δ in ppm, J in Hz) and 13C NMR (150 MHz, δ in ppm) data of compounds 1-4 in CDCl3. |


|
Download:
|
| Fig. 2. Selected HMBC (H → C) and ROESY (H ↔ H) correlations of compound 1. | |
Compound 2 was obtained as a white amorphous powder. Its molecular formula was assigned as C23H32O7 according to the HRESIMS ion peak at m/z 421.2247 [M+H]+ (calcd. m/z 421.2226, see Supporting information Fig. S14). The olefinic proton signal at δH 5.80 (s, 1H) in the 1H NMR spectrum, and the downfield carbon signals at δC 171.2, 170.0, 115.6, 107.7 in the 13C NMR spectrum (Table 1), indicated the presence of an α, β-unsaturated butenolide ring [13]. Besides, the 1H NMR spectrum exhibited signals for one methyl [δH 1.10 (d, 3H, J = 7.4 Hz)], three methoxyls [δH 3.17 (s, 3H), 3.32 (s, 3H) and 3.69 (s, 3H)], an oxygenated methylene [δH 3.30 (d, 1H, J = 11.2 Hz) and 3.90 (dd, 1H, J = 11.2, 2.7 Hz)], and one oxygenated methine [δH 4.74 (br s, 1H)]; and the 13C NMR spectrum exhibited 19 resonances attributed to three quaternary carbons (δC 174.7, 43.9 and 38.0), five methines (δC 102.4, 49.5, 42.3, 40.8 and 37.3), seven methylenes (δC 62.0, 37.4, 36.2, 35.6, 29.9, 23.8 and 21.0) and four methyls (δC 55.5, 51.6, 50.9 and 11.4) (Table 1). The 1H NMR and 13C NMR data were quite similar to those of caesalsappanin F [13]. After careful comparison, the ethoxy group in caesalsappanin F was replaced by a methoxy group in 2. In the HMBC spectrum, the correlations from OCH3-19 (δH 3.32, s) and H-5 (δH 1.68, m) to C-19 (δC 102.4), H-9 (δH 1.54, m) to C-20 (δC 62.0), and H-20 (δH 3.90, dd, J = 11.2, 2.7 Hz) to C-10 (δC 38.0), assigned the oxygenated methine and methylene as C-19 and C-20, respectively, and a methoxy group at C-19. The relative configuration of 2 was assigned by ROSEY spectrum. The NOE correlations of H-1α /H-5, H-5/H-9 and H-9/H3-17 indicated these protons and H3-17 were axial and α-oriented. H-19 was determined as α-oriented based on the correlation of H-19/H-3β in the ROESY spectrum. Therefore, the structure of 2 was established, named caesalsappanin O.
The molecular formula of compound 3 was determined by its HR-ESIMS (m/z 407.2081 [M+H]+, calcd. for C22H31O7, 407.2070, see Supporting information Fig. S21). The 1H NMR and 13C NMR data (Table 1) of 3 closely resembled those of caesalsappanin G [13]. The main difference was an additional methoxy group in 3. The HMBC correlation from the methoxy (δH 3.19) to C-12 (δC 108.0) suggested it was attached at C-12. H-19 was determined as α-oriented according to the NOE correlations of H-19/H-1β and H-19/H-2β. The NOE correlations between OCH3-12 and H3-17 indicated the methoxy group was α-faced. Thus, the structure of 3 was characterized, named caesalsappanin P.
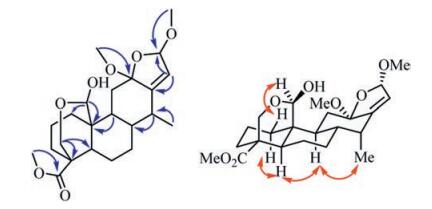
Compound 4 was obtained as a white amorphous powder. It molecular formula was deduced as C23H34O7 from the ion peak in the HR-ESIMS (m/z 445.2199 [M+Na]+, calcd. 445.2202, see Supporting information Fig. S28). The 1H NMR spectrum of 4 (Table 1) showed the presences of four methyls [δH 1.01 (d, 3H, J = 7.3 Hz), 3.18 (s, 3H), 3.50 (s, 3H) and 3.66 (s, 3H)], an oxygenated methylene [δH 3.67 (d, 1H, J = 11.8 Hz) and 4.33 (dd, 1H, J = 11.8, 2.6 Hz)], two oxygenated methines [δH 4.83 (d, 1H, J = 2.1 Hz) and 5.35 (d, 1H, J = 1.4 Hz)], and an olefinic proton [δH 5.58 (d, 1H, J = 1.3 Hz)]. The 13C NMR spectrum (Table 1) displayed 23 carbon signals attributed to one methyl (δC 12.2), three methoxyls (δC 50.3, 51.6 and 56.1), seven methylenes (δC 20.9, 23.9, 29.2, 35.7, 37.3, 38.7 and 61.5), seven methines (δC 35.9, 41.2, 41.7, 45.0, 97.2, 105.7 and 120.4), and five quaternary carbons (δC 38.3, 45.5, 110.2, 148.4 and 175.7). The NMR data of 4 showed high similarity with those of 3, except that an oxygenated methine (δH 5.35, d, J = 1.4 Hz/δC 105.7) and a methoxyl (δH 3.50, s/δC 56.1) were observed in 4 rather than the carbonyl group (δC 170.3) in 3. In the HMBC spectrum of 4 (Fig. 3), the correlations of H-16 (δH 5.35) with C-12 (δC 110.2), C-13 (δC 148.4) and C-14 (δC 35.9), and H-15 (δH 5.58) with C-8 (δC 41.2), C-12 (δC 110.2) and C-13 (δC 148.4), demonstrated their proximity as caesalminaxin H [14]. The correlations between H-20 and H-1β, as well as OCH3-12 and H3-17, in the ROESY spectrum indicated that H-20 and OCH3-12 were α-oriented (Fig. 3). OCH3-16 was determined as α-oriented according to the reference [14]. Thus, the structure of 4 was determined, named caesalsappanin Q.

|
Download:
|
| Fig. 3. Selected HMBC (H → C) and ROESY (H ↔ H) correlations of compound 4. | |
Compound 5 and phanginin B [11] were isolated as a pair of isomers, which were inseparable. To separate them, the mixture was methylated by methyl iodide. The reaction mixture was further purified by preparative HPLC to yield the methyl derivatives of compound 5 and phanginin B, 5a and phanginin D. The IR absorptions at 1722, 1239 and 1144 cm-1 suggested the presence of a carboxylic ester group in 5a. The 1H and 13C NMR spectra of 5a (Table 2) were quite similar to those of phanginin D [11]. The HMBC correlations of the oxymethine proton at δH 5.02 (s, 1H) with C-3 (δC 29.1), C-4 (δC 49.5), C-5 (δC 48.0) and C-20 (δC 67.4) assigned it as H-19. The relative configuration of 5a was confirmed through ROESY experiment. The NOE correlations between H-19 and H-6β suggested that H-19 was β-oriented. Thus, the structure of 5a was determined. Compound 5 was proposed as a demethylated derivative of 5a, and it was named as phanginin U.
|
|
Table 2 1H (600 MHz, δ in ppm, J in Hz) and 13C NMR (150 MHz, δ in ppm) data of compounds 5 and 5a in CDCl3. |

Five known diterpenoids were identified as tomocin B (6) [15], tomocin G (7) [15], phanginin J (8) [11], 20-nor-10α-hydroperoxyphanginin K (9) [16], and 19α-hydroxy-20-O-methylphanginin A (10) [16] by comparing their spectroscopic data with those reported in the literature.
Compounds 1-4 and 5a were tested for their cytotoxic activity against MCF-7 and HCT116 cancer cell lines. All of them showed moderate cytotoxic activity against two cell lines (Table 3). Especially, compound 4 showed inhibitory effect on MCF-7 (28.8 ± 0.5%) and HCT116 (46.1 ± 5.4%) cells at the concentration of 20mmol/L.
|
|
Table 3 Cytotoxicity of compounds 1–4 and 5a against MCF-7 and HCT116 cell lines. |

{kind=link}
{kind=link}
{kind=link}
3. Conclusion
Five new cassane-type diterpenoids, 11-oxo-phanginin A (1), caesalsappanins O-Q (2-4) and phanginin U (5), together with five known analogues (6-10), were isolated from the seeds of C. sappan. Compounds 1-3 and 5 possess a common cassane-type skeleton with a furano or an α, β-unsaturated butenolide ring, and compound 1 contains a rare carbonyl group at C-11. Compound 4 represents the first example of cassane-type diterpenoid with a 19, 20-epoxide linkage and a methoxy group at C-16 from C. sappan.
4. Experimental 4.1. General experimental proceduresOptical rotation data were obtained using an Autopol VI polarimeter. UV data were recorded with a Varian CARY 50 spectrophotometer. IR spectra were recorded on a PerkinElmer spectrum-100 FTIR spectrometer using KBr disks. NMR spectra were recorded on a Bruker Avance-600 (Bruker, Switzerland, 600 MHz for 1H NMR, 150 MHz for 13C NMR) NMR spectrometers with the chemical shift values presented as δ values having TMS as an internal standard. The coupling constants (J) were given in Hz. HR-ESIMS spectra were recorded on an LTQ-Orbitrap XL spectrometer. All solvents were analytical grade (TianJing Chemical Plant, Tianjing, China). Silica gel used for column chromatography and precoated silica gel GF254 plates used for TLC were produced by Qingdao Haiyang Chemical Co., Ltd. TLC spots were viewed at 254 nm and visualized by spraying with 1% vanillin in H2SO4. MCI gel (CHP20P, 75-150mm, Mitsubishi Chemical Industries Ltd.) and ODS C18 gel (50 mm, YMC, Kyoto, Japan) was used for column chromatography (CC). Preparative HPLC was performed on a Shimadzu LC-20AP instrument with a SPD-M20A PDA detector. Chromatographic separation was carried out on a C18 column (19 × 250 mm, 5mm, Waters, SunFireTM), using a gradient solvent system comprised of H2O (A) and CH3CN (B) at a flow rate of 10 mL/min.
4.2. Plant materialThe seeds of Caesalpinia sappan Linn. were collected in Baise City, Guangxi Province, People's Republic of China, and authenticated by Professor Jingui Shen from Shanghai Institute of Materia Medica, Chinese Academy of Sciences. A specimen was deposited at the herbarium of Institute of Chinese Medical Sciences, University of Macau (LL-20140601).
4.3. Extraction and isolationThe air-dried seeds of C. sappan (15.1 kg) were ground into powder and extracted with petroleum ether for three times to remove lipids. Then the residues were extracted three times with 80% ethanol under reflux and evaporated to afford a crude extract (500.4 g). The extract was suspended in 2.0 L H2O and extracted with chloroform (1.5 L × 3) to yield a chloroform-soluble extract (370.0 g). The chloroform extract was subjected to CC over silica gel and eluted with petroleum ether-acetone (19:1, 15:1, 10:1, 7:1, 5:1, 1:1, v/v), to yield ten major fractions (C1 to C10). Fraction C1 was were subjected to an MCI gel column eluted with CH3OH-H2O (60:40, 70:30, 80:20, 90:10, 100:0, v/v) to yield subfractions C1A-C1J. Subfraction C1C was purified by preparative HPLC eluted with gradient H2O/CH3CN (2:3 to 0:1, v/v) to obtain compound 9 (1.3 mg). Fraction C6 (19.6 g) was subjected to CC over silica gel and eluted with petroleum ether-acetone (60:1, 40:1, 30:1, 20:1, 10:1, 0:1, v/v) to yield subfractions C6A-C6F. Fraction C6C was subjected to a silica gel column eluted with gradient petroleum etheracetone (30:1 to 10:1, v/v), and further purified using preparative HPLC to obtain 2 (3.2 mg), 3 (1.0 mg), 4 (1.2 mg) and 10 (2.6 mg). C6D was subjected to an ODS column eluted with CH3OH/H2O (40:60, 60:80, 80:20, 90:10, 100:0, v/v), followed by preparative HPLC to yield 1 (11.2 mg), 6 (0.8 mg) and 7 (8.9 mg). C6E was further chromatographed over silica gel eluted with gradient petroleum ether-acetone (30:1, 20:1, 10:1, v/v) to obtain 5 (120 mg) and 8 (1.1 mg).
11-Oxophanginin A (1): White amorphous powder; [α]D20 + 18.7 (c 0.1, CH3OH); UV (CH3OH) λmax (log ε) 271 nm (3.93); IR νmax (KBr) 3431 (strong, broad), 2928, 1727, 1667, 1430, 1258, 1101, 1056 cm-1; 1H NMR and 13C NMR data see Table 1; HR-ESIMS m/z 397.1634 [M+Na]+ (calcd. for C21H26O6Na, 397.1627).
Caesalsappanin O (2): White amorphous powder; [α]D20 -6.8 (c 0.1, CH3OH); UV (CH3OH) λmax (log ε) 209 nm (3.76); IR νmax (KBr) 3430 (strong, broad), 2924, 1768, 1734, 1631, 1383, 1118, 1053 cm-1; 1H NMR and 13C NMR data see Table 1; HR-ESIMS m/z 421.2247 [M +H]+ (calcd. for C23H33O7, 421.2226).
Caesalsappanin P (3): White amorphous powder; [α]D20 -124 (c 0.1, CH3OH); UV (CH3OH) λmax (log ε) 213 nm (3.53); IR νmax (KBr) 3448 (strong, broad), 2992, 1757, 1643, 1262, 1099, 1043 cm-1; 1H NMR and 13C NMR data see Table 1; HRSEIMS m/z 407.2081 [M+H]+ (calcd. for C22H31O7, 407.2070).
Caesalsappanin Q (4): White amorphous powder; [α]D20 + 17.4 (c 0.1, CH3OH); UV (CH3OH) λmax (log ε) 203 nm (4.07); IR νmax (KBr) 3431 (strong, broad), 2926, 1729, 1632, 1383, 1101, 1058 cm-1; 1H NMR and 13C NMR data see Table 1; HR-ESIMS m/z 445.2199 [M +Na]+ (calcd. for C23H34O7Na, 445.2202).
Methylphanginin U (5a): White amorphous powder; [α]D20 + 35.0 (c 0.1, CH3OH); UV (CH3OH) λmax (log ε) 217 nm (3.77); IR νmax (KBr) 3443 (strong, broad), 2926, 1731, 1632, 1383, 1254, 1101, 1090 cm-1; 1H NMR and 13C NMR data see Table 2; HRESIMS m/z 375.2162 [M+H]+ (calcd. for C22H31O5, 375.2171).
4.4. Methylation of mixture of phanginins B and U (5)The mixture of phanginins B and U (5) (58 mg, 0.16 mmol) was dissolved in dehydrated acetone (3 mL), and the solution was treated with powdered potassium carbonate (0.6 g, 4.3 mmol) and methyl iodide (0.6 mL, 9.6 mmol). After stirred at room temperature for 17 h under an N2 atmosphere, the reaction mixture was evaporated under reduced pressure to remove the solvent. The residue was suspended in the ice-cooled H2O (10 mL) and extracted with EtOAc (3 ×10 mL). The combined organic phase was washed with satd aq NaCl (3 ×15 mL), dried over MgSO4, filtered, and evaporated in vacuo to give an oily residue, which by preparative HPLC (CH3OH/H2O = 85:15) gave 5a (27 mg, 45%).
4.5. Cytotoxicity bioassaysThe isolates were evaluated their cytotoxic effects against MCF-7 and HCT116 cancer cell lines using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) method, as described previously [8]. The human breast cancer MCF-7 cells and human colon cancer HCT116 cells were acquired from American Type Culture Collection (Manassas, VA, USA). MCF-7 and HCT116 cell lines were cultured in DMEM and RPMI1640 medium, respectively, supplemented with 10% (v/v) FBS and 1% (v/v) PenicillinStreptomycin in a standard humidified incubator with 5% CO2. Cells were dispensed in a 96 well sterile microplate (1 ×104 cells/ well), and incubated with series different concentrations of tested compound or Doxorubicin® (positive control) for 48 h at 37 ℃. After incubation, MTT reagent (1 mg/mL) was added to each well and then the 96-well plate was incubated for an additional 4 h. The purple formazan dye crystals were solubilized by the addition of 100mL of DMSO. The absorption at 570 nm was measured using a microplate reader (Perkin Elmer, 1420 Multilabel Counter Victor 3, Wellesley, MA, USA).
Competing financial interestsThe authors declare no competing financial interests.
AcknowledgmentsFinancial support by Science and Technology Development Fund, Macao S.A.R (No. FDCT 120/2013/A3) and the Research Fund of University of Macau (Nos. MYRG2014-00020-ICMS-QRCM and MYRG2015-00153-ICMS-QRCM) are gratefully acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.04.023.
| [1] | Editorial Board of Flora of China, vol. 39, Science Press, Beijing, 1988 pp.105. |
| [2] | S. Badami, S.R. Rai, S. Moorkoth, S. Rajan, B. Suresh. Pharmacognostical evaluation of Caesalpinia sappan heartwood. Anc. Sci. Life 23 (2003) 100–107. |
| [3] | D. Wang, C. Chen, Y. Zhou. The research progress of clinical pharmacology and chemical constituents from Caesalpinia sappan L. Inf. Tradit. Chin. Med. 20 (2003) 15–16. |
| [4] | M.A. Gómez-Hurtado, F.E. Álvarez-Esquivel, G. Rodríguez-García, et al., Cassane diterpenes from Caesalpinia platyloba. Phytochemistry 96 (2013) 397–403. DOI:10.1016/j.phytochem.2013.09.028 |
| [5] | W. Pranithanchai, C. Karalai, C. Ponglimanont, S. Subhadhirasakul, K. Chantrapromma. Cassane diterpenoids from the stem of Caesalpinia pulcherrima. Phytochemistry 70 (2009) 300–304. DOI:10.1016/j.phytochem.2008.12.006 |
| [6] | O. Yodsaoue, C. Karalai, C. Ponglimanont, S. Tewtrakul, S. Chantrapromma. Potential anti-inflammatory diterpenoids from the roots of Caesalpinia mimosoides Lamk. Phytochemistry 71 (2010) 1756–1764. DOI:10.1016/j.phytochem.2010.06.016 |
| [7] | J.L.B. Zanin, B.A. De Carvalho, P. Salles Martineli, et al., The genus Caesalpinia L. (Caesalpiniaceae):phytochemical and pharmacological characteristics. Molecules 17 (2012) 7887–7902. |
| [8] | H. Bao, L.L. Zhang, Q.Y. Liu, et al., Cytotoxic and pro-apoptotic effects of cassane diterpenoids from the seeds of Caesalpinia sappan in cancer cells. Molecules 21 (2016) 791. |
| [9] | Y.J. Xu, J. Zhang, C.P. Tang, Y. Ye. A new diterpenoid from the seeds of Caesalpinia sappan Linn. Rec. Nat. Prod. 7 (2013) 124–128. |
| [10] | F. Xiao, C.P. Tang, C.Q. Ke, S. Yao, Y. Ye. Rearranged diterpenoids from the seeds of Caesalpinia sappan. Chin. Chem. Lett. 27 (2016) 1751–1754. DOI:10.1016/j.cclet.2016.04.022 |
| [11] | H. Bao, Q.W. Zhang, Y. Ye, L. Lin. Naturally occurring furanoditerpenoids:distribution, chemistry and their pharmacological activities. Phytochem. Rev. 16 (2016) 235–270. |
| [12] | O. Yodsaoue, S. Cheenpracha, C. Karalai, et al., Phanginin A-K, diterpenoids from the seeds of Caesalpinia sappan Linn. Phytochemistry 69 (2008) 1242–1249. DOI:10.1016/j.phytochem.2007.11.013 |
| [13] | G.X. Ma, H.F. Wu, D.L. Chen, et al., Antimalarial and antiproliferative cassane diterpenes of Caesalpinia sappan. J. Nat. Prod. 78 (2015) 2364–2371. DOI:10.1021/acs.jnatprod.5b00317 |
| [14] | Y. Zheng, S.W. Zhang, H.J. Cong, Y.J. Huang, L.J. Xuan. Caesalminaxins A-L, cassane diterpenoids from the seeds of Caesalpinia minax. J. Nat. Prod. 76 (2013) 2210–2218. DOI:10.1021/np400545v |
| [15] | H.X. Nguyen, N.T. Nguyen, P.H. Dang, et al., Cassane diterpenes from the seed kernels of Caesalpinia sappan. Phytochemistry 122 (2016) 286–293. DOI:10.1016/j.phytochem.2015.12.018 |
| [16] | Z.T. Deng, J. Su, L.F. Ding, et al., Six new cassane diterpenoids from the seeds of Caesalpinia sappan. Phytochem. Lett. 16 (2016) 207–212. DOI:10.1016/j.phytol.2016.04.014 |