 2017, Vol. 28
2017, Vol. 28 



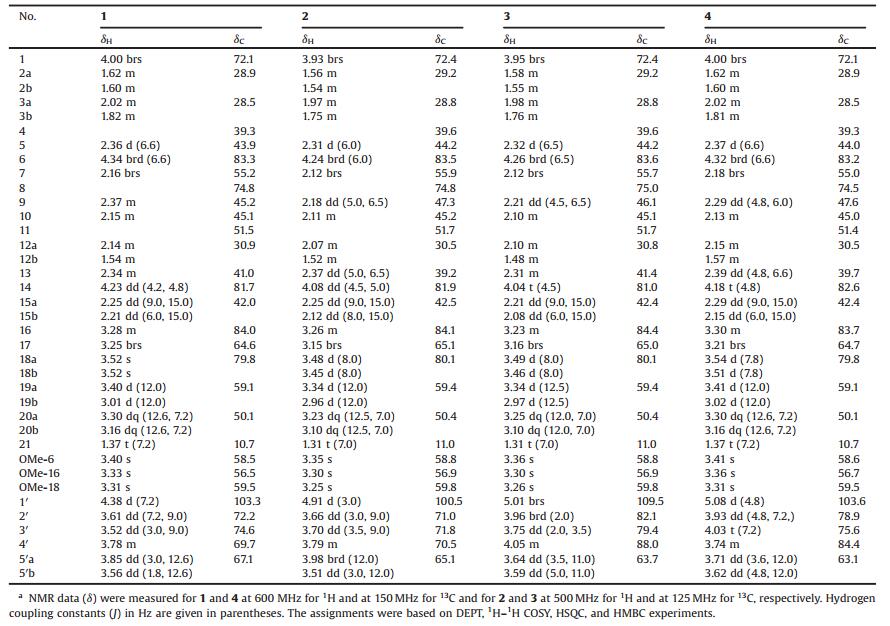
The lateral roots of Aconitum carmichaelii Debx. (Ranunculaceae), named "fu zi" in Chinese, is an indispensable herbal medicine for the treatment of various diseases, such as cardianeuria, arrhythmia, neuralgia, rheumatalgia, and inflammations, in oriental countries [1-4]. Chemical and pharmacological investigations have resulted in isolation of more than a hundred of compounds from extracts of the principal and lateral roots (tubers) of A. carmichaelii, of which aconitane-type C19-diterpenoid alkaloids were reported as the main active constituents [2-12]. However, in previous chemical studies organic solvents (benzene, CHCl3, methanol, and ethanol) were mainly applied for extracting the plant materials including raw and prepared "fu zi" [2-11]. These extraction procedures differ from a practical utilization by decocting with water. Therefore, as part of a program to systematically study the chemical diversity of traditional Chinese medicines and their biological effects [13-33], we investigated an aqueous decoction of the raw lateral roots of A. carmichaelii. As results of our investigation, four new hetisan-type, three new napeline-type, and a new arcutine-type C20-diterpenoid alkaloids, twenty-one new aconitane-type C19-diterpenoid alkaloids, two new 2-(quinonylcarboxamino)benzoates, and seven new aromatic acid derivatives, along with solvent-/base-/acid-dependent transformation and equilibration between alcohol iminium and aza acetal forms of the napeline-type C20-diterpenoid alkaloids, have been reported [34-38]. Described herein are isolation and structural characterization of four unprecedented aconitane-type C19-diterpenoid alkaloid glycosides with isomeric L-arabinosyls (1-4) (Fig. 1), from the same decoction.

|
Download:
|
| Fig. 1. Structures of 1-4. | |
2. Results and discussion
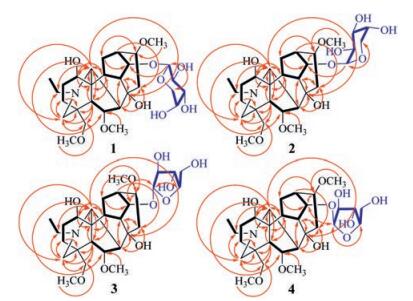
Compound 1 was isolated as a colorless gum with [α]D20 -11.6 (c 0.69, MeOH). Its IR spectrum exhibited absorption bands assignable to hydroxyl (3356 cm-1) groups. The molecular formula of 1 was elucidated as C29H47NO10 by (+)-HR-ESIMS at m/z 570.3283 [M+H]+ (calcd. for C29H48NO10, 570.3273), combined with NMR spectroscopic data (Table 1). The 1H NMR spectrum of 1 in MeOH-d4 showed resonances attributable to an N-CH2CH3 unit at δH 3.30 and 3.16 (dq, 1H each, J = 12.6 and 7.2 Hz, H2-20) and 1.37 (t, 3H, J = 7.2 Hz, H3-21); three methoxy groups at δH 3.40 (s, OCH3-6), 3.33 (s, OCH3-16), and 3.31 (s, OCH3-18); four oxymethines at δH 4.34 (brd, J = 6.6 Hz, H-6), 4.23 (dd, J = 4.2 and 4.8 Hz, H-14), 4.00 (brs, H-1), and 3.28 (m, H-16); one nitrogen-bearing methine at δH 3.25 (brs, H-17); an oxymethylene at δH 3.52 (2H, s, H2-18); and a nitrogen-bearing methylene at δH 3.40 and 3.01 (d, 1H each, spectroscopic data analysis (Table 1). In the 1H-1H COSY spectrum of 1, homonuclear vicinal coupling correlations of H-1/H2-2/H2-3, H-5/H-6/H-7, H-14/H-9/H-10/H2-12/H-13/H-14, H2-15/H-16, and H2-20/H3-21 revealed five vicinal spin systems of the aglycone moiety (Fig. 2, thick lines). In the HMBC spectrum of 1, two-and three-bond correlations (Fig. 2, red arrows) from H2-3 to C-4 and C-19; from H-5 to C-4, C-18, and C-19; from H2-18 to C-3, C-4, C-5, and C-19; and from H2-19 to C-3, C-4, C-5, and C-18 indicated a J = 12.0 Hz, H2-19); as well as partially overlapped resonances due to four aliphatic methylenes (H2-2, H2-3, H2-12, and H2-15) and five aliphatic methines (H-5, H-7, H-9, H-10, and H-13) between δH 2.37 and 1.54. In addition, the spectrum showed typical signals for a pentose moiety, consisting of four oxygen-bearing methines at δH 4.38 (d, J = 7.2 Hz, H-1'), 3.61 (dd, J = 7.2 and 9.0 Hz, H-2'), 3.52 (dd, J = 3.0 and 9.0 Hz, H-3'), and 3.78 (m, H-4') and an oxygen-bearing methylene at δH 3.85 (dd, J = 3.0 and 12.6 Hz, H-5'a) and 3.56 (dd, J = 1.8 and 12.6 Hz, H-5'b). The 13C NMR and DEPT spectra showed 29 carbon signals (Table 1) corresponding to the above units and three quaternary carbons including an oxygen-bearing carbon at δC 74.8 (C-8). As compared with those of the alkaloids previously isolated from "fu zi", the spectroscopic data suggest that 1 is an unprecedented glycosidic C19-diterpenoid alkaloid [12, 34-38], of which the structure was elucidated by 2D NMR data analysis.
|
|
Table 1 NMR spectroscopic data for 1-4 in MeOH-d4.a |
{kind=link}

|
Download:
|
| Fig. 2. Main 1H–1H COSY (thick lines) and HMBC correlations (red arrows, from 1H to 13C) of 1-4. | |
{kind=link}
The hydrogen resonances and corresponding hydrogen-bearing carbon resonances in the NMR spectra were assigned by HSQC spectroscopic data analysis (Table 1). In the 1H-1H COSY spectrum of 1, homonuclear vicinal coupling correlations of H-1/H2-2/H2-3, H-5/H-6/H-7, H-14/H-9/H-10/H2-12/H-13/H-14, H2-15/H-16, and H2-20/H3-21 revealed five vicinal spin systems of the aglycone moiety (Fig. 2, thick lines). In the HMBC spectrum of 1, two-and three-bond correlations (Fig. 2, red arrows) from H2-3 to C-4 and C-19; from H-5 to C-4, C-18, and C-19; from H2-18 to C-3, C-4, C-5, and C-19; and from H2-19 to C-3, C-4, C-5, and C-18 indicated a linkage of the quaternary C-4 with C-3, C-5, C-18, and C-19. The HMBC correlation from H-7 to C-8, C-9, and C-15; from H-9 to C-7, C-8, and C-15; from H2-15 to C-7, C-8, and C-9; together with their chemical shifts, demonstrated that the oxygen-bearing quaternary C-8 was linked to C-7, C-9 and C-15. The HMBC correlation from H-1 to C-10, C-11, and C-17; from H-5 to C-10, C-11, and C-17; from H-10 to C-1, C-5, C-11, and C-17; and from H-17 to C-5, C-10, C-11 indicated that the quaternary C-11 connected to C-1, C-5, C-10 and C-17. The connection between C-13 and C-16 was established by the HMBC correlations from H2-12 to C-14 and C-16, from H-13 to C-15, from H-14 to C-16, and from H-16 to C-12 and C-14, though the vicinal coupling correlation between H-13 and H-16 was not observed in the 1H-1H COSY spectrum possibly due to a perpendicular dihedral angle between the two hydrogens. In addition, the HMBC correlations from H-6 to C-17; from H-17 to C-6, C-8, C-19, and C-20; from H2-19 to C-17 and C-20; from H2-20 to C-17 and C-19; together with their chemical shifts, demonstrated that C-17 connected with C-7 and via the nitrogen atom to both C-19 and C-20. The HMBC correlations from OCH3-6 to C-6, from OCH3-16 to C-16, and from OCH3-18 to C-18 located the three methoxy groups at the corresponding carbons. Furthermore, the 1H-1H COSY spectrum of 1 exhibited the correlations of H-1'/H-2'/ H-3'/H-4'/H2-5'. This, combined with the HMBC correlation from H-1' to C-5' and C-14, from H2-5' to C-1', and from H-14 to C-1' demonstrated that there was a pentopyranosyloxy unit at C-14. The hydroxyl group at C-8 was deduced from the chemical shift of this carbon and the molecular formula requirement. Accordingly, the planar structure of 1 was determined as shown, wherein the planar structure of the aglycone is identical to the co-occurring neoline [39, 40].
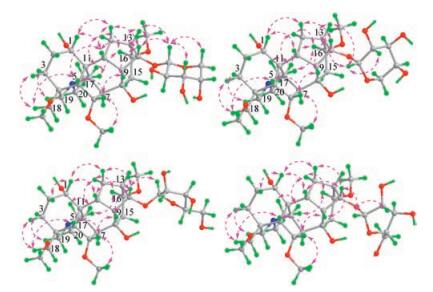
In the NOESY spectrum of 1, the NOE correlations between H-1 with H-10 and H-12a, between H-3a and H2-18, between H-5 with H-10 and H2-18, between H-6 and H-9, between H-10 with H-12a and H-14, and between H-14 with H-9 and H-13 indicated that these hydrogens were oriented on the same side of the ring system (Fig. 3). The NOE correlations between H-3b with H-19b, between H-16 with H-12b and H3-21, and between H-17 with H-12b and H3-21 revealed that these hydrogens were oriented on the other side of the ring system. These NOE correlations indicated that the relative configuration of the aglycone is identical to neoline [39, 40]. Additionally, the NOE correlations between H-1' with H-3' and H-5'b, together with the coupling constant values of J1', 2' (7.2Hz) and J3', 4' (3.0Hz), demonstrated that the pentopyranosyloxy in 1 is an α-arabinopyranosyloxy [41], while the NOE correlations between H-1' and H-14 confirmed the location of the sugar unit. Altogether, 1 was deduced as neoline 14-O-α-arabinopyranoside, of which the absolute configuration was further assigned by acid hydrolysis. The aglycone and sugar, which were isolated from the hydrolysate of 1 by CC over Sephadex LH-20 (MeOH), displayed the 1H NMR spectroscopic features (Figs. S13-S17 in Supporting information) and the specific rotation values {aglycone:[α]D20 +21.8 (c 0.05, MeOH); sugar: [α]D20 +104.8 (c 0.01, H2O)} well consistent with those of the authentic samples of neoline {[α]D20 +21.6 (c 0.16, MeOH)} and L-arabinose {[α]D20 +113.3 (c 0.29, H2O)}. Especially the absolute configuration of neoline was previously determined by chemical transformation and X-ray crystallography [42-44]. Thus, the structure of compound 1 was determined as neoline 14-O-α-L-arabinopyranoside and named aconicarmichoside A.

|
Download:
|
| Fig. 3. Main NOESY correlations (pink dashed double arrows) of 1-4. | |
{kind=link}
Compound 2, a colorless gum with [α]D20 +46.3 (c 0.54, MeOH), exhibited spectroscopic features similar to those of 1. However, as compared with the NMR spectroscopic data of 1, the H-14 and C-13 resonances in 2 were shielded by ΔδH -0.15 and ΔδC -1.8, respectively, whereas the anomeric proton resonance (H-1') was significantly deshielded by ΔδH +0.53 (Table 1). Additionally, the coupling constants of H-1' (J1', 2') was changed from 7.2Hz in 1 into 3.0Hz in 2. This suggests that the anomeric carbon (C-1') of the arabinopyranosyl in 2 has a b-configuration instead of the a-configuration in 1. The suggestion was supported by shielded shifts of the C-1', C-3', and C-5' resonances in the 13C NMR of 2 by ΔδC -2.8, -2.8, and -2.0, respectively, as compared with those of 1. This was further confirmed by 2D NMR data analysis and acid hydrolysis of 2 (Figs. 2 and 3). Especially, the homonuclear vicinal coupling correlations of H-1'/H-2'/H-3'/H-4'/H2-5' in the 1H-1H COSY spectrum and the long-range heteronuclear correlation from H-1' to C-5', from H-5' to C-1', from H-1' to C-14 and from H-14 to C-10 in the HMBC spectrum, together with the coupling constants of J2', 3' (9.0Hz) and J3', 4' (3.5Hz), proved the presence and location of the β-arabinopyranosyloxy in 2. Using the same method as described for 1, neoline {[[α]D20 +20.9 (c 0.08, MeOH)} and L-arabinose {[α]D20 +106.2 (c 0.03, H2O)} was isolated and identified from the hydrolysate of 2. Therefore, the structure of compound 2 was determined as neoline 14-O-β-L-arabinopyranoside and named aconicarmichoside B.
Compound 3, a colorless gum with [α]D20 -20.6 (c 0.31, MeOH), is another isomer of 1 and 2 as indicated by spectroscopic data. Comparison of the NMR spectroscopic data of 1-3 (Table 1) demonstrated that the anomeric hydrogen and carbon of the pentosyl in 3 resonated with deshielded chemical shifts at δH 5.01 (H-1') and δC 109.5 (C-1'), respectively, while the H-1' resonance appeared as a singlet. This suggests that the pentosyl in 3 is an α-arabinofuranosyl, which was confirmed by 2D NMR data analysis of 3 (Figs. 2 and 3). Especially the 1H-1H COSY crosspeaks of H-1'/H-2'/H-3'/H-4'/H2-5' and the HMBC correlations from H-1' to C-4' and from H-4' to C-1', as well as the coupling constants of J2', 3' (2.0Hz) and J3', 4' (3.5Hz), indicated the presence of α-arabinofuranosyl in 3. Meanwhile, the HMBC correlations from H-1' to C-14 and from H-14 to C-1' verified the location of the sugar unit at C-14 in 3. Moreover, neoline {[α]D20 +22.3 (c 0.08, MeOH)} and L-arabinose {[α]D20 +107.6 (c 0.04, H2O)} were isolated and identified from the hydrolysate of 3. Thus, the structure of compound 3 was determined as neoline 14-O-α-L-arabinofuranoside and named aconicarmichoside C.
Compound 4 was isolated as a colorless gum with [α]D20 +37.1 (c 0.50, MeOH). The spectroscopic data of 4 indicated that this compound is one more isomer of 1-3, in which the anomeric proton (H-1') had a more deshielded chemical shift (δH 5.08) and a coupling constant value of 4.8 Hz (J1', 2') (Table 1). This, combined with 2D NMR data analysis (Figs. 2 and 3), suggested replacement of the α-arabinofuranosyl in 3 by a β-L-arabinofuranosyl in 4. The suggestion was proved by acid hydrolysis of 4. From the hydrolysate of 4, neoline {[α]D20 +20.7 (c 0.07, MeOH)} and Larabinose {[α]D20 +110.1 (c 0.03, H2O)} were isolated and identified as the sole aglycone and sugar. Therefore, the structure of compound 4 was determined as neoline 14-O-β-L-arabinofuranoside and named aconicarmichoside D.
Interestingly, the specific rotations of 1 and 3 with the α-L-arabinosyls are reversed to those of 2 and 4 with the β-L-arabinosyls, though both the aglycone and sugar have the positive specific rotations. This suggests that the specific rotations of the neoline 14-O-β-L-arabinosides are dominated by the C-1' configurations of the L-arabinosyls.
Because trifluoroacetic acid (TFA) was used in the final HPLC purification steps, the basicity of the diterpenoid alkaloids suggests that 1-4 were possibly obtained as trifluoroacetates or complexes [34, 36-38, 45-47]. However, in the ESIMS of 2 and 4, ion peaks at m/z 592 (positive mode) and 604 and/or 568 (negative mode), ascribable to [alkaloid + Na]+ and [alkaloid + HCl-H]- and/ or [alkaloid-H]-, respectively, supported the occurrence of the free bases and/or an equilibration of interactions between the alkaloids and acids (Figs. S18 and S44 in Supporting information). Although the 13C resonances of TFA- (or TFA) were not observed in the 13C NMR spectra of 1-4 (Figs. S7, S23, S37, and S51 in Supporting information), the 19F NMR spectra displayed a 19F resonance attributable to TFA- (or TFA) around δF -76.5, indicating the presence of TFA- (or TFA) in the samples. Subsequent quantitative 19F NMR analysis of 1-4, using benzene (C6H6) and hexafluorobenzene (C6H6) as internal standards, determined that approximate ratios of the alkaloids and TFA in the samples were 1:1.2-1:1.7 (Figs. S59-S66 in Supporting information). The absence of the 13C resonances of TFA- (or TFA) in the 13C NMR spectra should be due to comprehensive reasons, including spin couplingsplit of the 13C resonances by the 19F nucleus, conjugation between different carboxylic species of TFA, and interactions of the carboxylic species of TFA with the alkaloid and solvent in the solution state. This is similar to that observed in our previous studies [34, 36-38]. Since TFA came from the experimental procedures and since a variety of acids are exist in the bio-system, under in vivo hydrophilic conditions these diterpenoid alkaloids should interact with the acids to increase their solubility, bioavailability, and transportations, as well as to play biological functions. In addition, in specific and/or local alkali microenvironments of the bio-systems the free base forms, along with potential equilibrations of interactions with the acids, would also play potential roles.
In the in vitro bioassays, compounds 1 and 2 inhibited lipopolysaccharide (LPS)-induced NO production in cultivated murine microglial cell line BV-2 with 28.3% and 32.6% inhibitions at 10mmol/L, respectively, while the positive control curcumin gave 87.3% inhibition at the same concentration [48]. Compound 2 also showed antiviral activity against the influenza virus A/ Hanfang/359/95 (H3N2) with IC50 and SI values of 33.3mmol/L and 3.0, respectively, the positive control, ribavirin, gave IC50 = 1.71 mmol/L and SI = 680.8 [49]. This indicates that isomerization of the arabinosyl plays a role for the activities. Other assays, including influence onsynchronized Ca2+ oscillation in cultivated cardiac muscle cell, activity against KCNQ2 potassium channel in CHOK1 cells, protection against L-glutamic acid-induced SK-N-SH neuroblastoma cell damage, inhibition against Fe2+-cysteine induced rat liver microsomal lipid peroxidation, antiviral activity against Coxsackie virus B3 (CVB3), and cytotoxicity against several human cancer cell lines were also performed in this study. However, 1-4 were inactive at a concentration of 10mmol/L in each assay.
3. ConclusionFour aconitine-type C19-diterpenoid alkaloids 1-4 were isolated and characterized from the aqueous extract of the lateral roots of A. carmichaelii ("fu zi"). Although a great number of diterpenoid alkaloids, covering C20-, C19-, and C18-categories, have been isolated from plant species of the Ranunculaceae, Rosaceae, Asteraceae, Garryaceae, Escalloniaceae, and Polygonaceae families during the past century, no diterpenoid alkaloid glycoside was reported as a natural or synthetic product [50-53]. This is abnormal because glycosidation of the free hydroxyl group(s) in many structures of the diterpenoid alkaloids would occur as commonly observed in the other types of natural products. Therefore, the discovery of this study amends and proves the natural occurrence of diterpenoid alkaloid glycosides, while 1-4 are the first examples of the unprecedented glycosidic diterpenoid alkaloids. In addition, 1-4 represent also the first examples that the glycosidic natural products with the four isomeric L-anabinosyls are simultaneously isolated from the same plant species. Although weak inhibitory activities against NO-production and influenza virus A/Hanfang/359/95 (H3N2) were found for 1 and/or 2, the potential activities of the unique C19-diterpenoid alkaloid isomers and contributions to the clinic effects of "fu zi" are still expected from future evaluations with an assistance of semi/total synthesis to obtain enough amount of the samples.
4. Experimental 4.1. General experimental proceduresOptical rotations were measured on a P-2000 polarimeter (JASCO, Tokyo, Japan). UV spectra were recorded on a V-650 spectrometer (JASCO). CD spectra were measured on a JASCO J-815 CD spectrometer (JASCO). IR spectra were recorded on a Nicolet 5700 FT-IR Microscope spectrometer (FT-IR Microscope Transmission) (Thermo Electron Corporation, Madison, WI, USA). 1D and 2D NMR spectra were obtained at 600 or 500 MHz for 1H, 150 or 125 MHz for 13C, respectively, on a SYS 600 MHz, an Inova 500 MHz (Varian Associates Inc., Palo Alto, CA, USA), a Bruker 600 MHz (Bruker Corp., Karlsruhe, Germany), or a WNMR-I 500 MHz (Wuhan Zhongke Niujin Magnetic Resonance Technology Co., Ltd., Wuhan, China) spectrometer, with TMS or solvent peaks as references. ESIMS and HR-ESIMS data were obtained on Agilent 1100 Series LC-MSD-Trap-SL and Agilent 6520 Accurate-Mass QTOFL CMS spectrometers (Agilent Technologies, Ltd., Santa Clara, CA, USA), respectively. Column chromatography (CC) was performed with silica gel (200-300 mesh, Qingdao Marine Chemical Inc., China), reversed phase C-18 silica gel (W. R. Grace & Co., Maryland, USA), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden). HPLC separation was performed on a system consisting of a Waters 600 controller, a Waters 600 pump, and a Waters 2487 dual absorbance (Waters Corporation, Milford, MA, USA) or a Smartline RI (Knauer, Berlin, Germany) detector, using an Ultimate XB-Phenyl column (250 mm × 10 mm i.d.) packed with phenyl-silica gel (5mm) (Welch, shanghai, China). TLC was conducted on precoated silica gel GF254 plates. Spots were visualized under UV light (254 or 365 nm) or by spraying with 7% H2SO4 in 95% EtOH followed by heating or with a Dragendorff's reagent. Unless otherwise noted, all chemicals were obtained from commercially available sources and were used without further purification.
4.2. Plant material 4.3. Extraction and isolationFor extraction and preliminary fractionation of the extract, see refs 34-38. Fraction C2-2 (200 g) was separated by CC over Sephadex LH-20 (CHCl3-MeOH, 1:1) yielded C2-2-1-C2-2-8. Fraction C2-2-4 (9.5 g) was chromatographed over silica gel (150 g) eluting with a gradient of petroleum ether-Me2CO-diethylamine (5:2:1-2:2:1) to give C2-2-4-1-C2-2-4-7. Fraction C2-2-4-6 (2.65 g) was separated by CC over silica gel, eluting with a gradient of CHCl3 (saturated with ammonia water)-MeOH (20:1-5:1), to give C2-2-4-6-1-C2-2-4-6-11, of which C2-2-4-6-6 (1.5 g) was further fractionated by CC over reversed phase C-18 silica gel (30-50% MeOH in H2O) to give C2-2-4-6-6-1-C2-2-4-6-6-3. Fraction C2-2-4-6-6-1 (1.2 g) was subjected to preparative TLC [CHCl3 (saturated with ammonia water)-MeOH, 5:1] to yield C2-2-4-6-6-1-1 and C2-2-4-6-6-1-2. Further separation of C2-2-4-6-6-1-1 (0.9 g) by reversed phase HPLC (Ultimate XB-Phenyl column, 15% MeCN in H2O containing 0.1% TFA, 2 mL/min) yielded C2-2-4-6-6-1-1-1-C2-2-4-6-6-1-1-5, of which C2-2-4-6-6-1-1-5 was purified by reversed phase HPLC using the same column and the mobile phase of 42% MeOH in H2O containing 0.1% TFA and the flow rate 2 mL/min to afford 4 (6.5 mg, tR = 25 min). Fraction C2-2-4-6-6-2 (0.3 g) was chromatographed over silica gel eluting with CHCl3 (saturated with ammonia water)-MeOH (5:1), to give C2-2-4-6-6-2-1-C2-2-4-6-6-2-5, of which C2-2-4-6-6-2-4 (39 mg) was separated by HPLC (Ultimate XB-Phenyl column, 45% MeOH in H2O containing 0.1% TFA, 2 mL/min) to yield C2-2-4-6-6-2-4-1 and C2-2-4-6-6-2-4-2, which were separately purified by HPLC using the same column. Using the mobile phase of 35% MeOH in H2O containing 0.1% TFA (2 mL/min), 3 (4 mg, tR = 49 min) was obtained from the former, and using the mobile phase of 20% MeOH in H2O containing 0.1% TFA (2 mL/min), 2 (7 mg, tR = 44 min) was obtained from the latter. Fraction C2-2-4-6-7 (110 mg) was separated by preparative TLC [CHCl3 (saturated with ammonia water)-MeOH (5:1)] to afford C2-2-4-6-7-1-C2-2-4-6-7-3, of which C2-2-4-6-7-1 (20 mg) was purified by reversed phase HPLC (Ultimate XBPhenyl column, 19% acetonitrile in H2O containing 0.1% TFA, 2 mL/ min) to yield 1 (9 mg, tR = 24 min).
Aconicarmichoside A (1): Colorless gum; [α]D20 -11.6 (c 0.69, MeOH); IR νmax 3356, 2935, 1678, 1448, 1291, 1201, 1128, 1010, 942, 836, 801, 721 cm-1; 1H NMR (MeOH-d4, 600 MHz) data, see Table 1; 13C NMR (MeOH-d4, 150 MHz) data, see Table 1; (+)-ESIMS m/z 570 [M+H]+; (+)-HR-ESIMS m/z 570.3283 [M+H]+ (calcd. for C29H48NO10, 570.3273).
Aconicarmichoside B (2): Colorless gum; [α]D20 +46.3 (c 0.54, MeOH); IR νmax 3369, 2935, 1678, 1451, 1356, 1202, 1133, 1082, 1011, 938, 886, 837, 800, 721 cm-1; 1H NMR (MeOH-d4, 500 MHz) data, see Table 1; 13C NMR (MeOH-d4, 125 MHz) data, see Table 1; (+)-m/ z ESIMS 592 [M+Na]+, 570 [M+H]+; (-)-ESIMS m/z 604 [M+HCl-H]-; (+)-HR-ESIMS m/z 570.3273 [M+H]+ (calcd. for C29H48NO10, 570.3273).
Aconicarmichoside C (3): Colorless gum; [α]D20 -20.6 (c 0.31, MeOH); IR νmax 3393, 2924, 2851, 1679, 1438, 1352, 1205, 1134, 1076, 1048, 984, 962, 841, 802, 724 cm-1; 1H NMR (MeOH-d4, 500 MHz) data, see Table 1; 13C NMR (MeOH-d4, 125 MHz) data, see Table 1; (+)-ESIMS m/z 570 [M+H]+; (+)-HR-ESIMS m/z 570.3268 [M +H]+ (calcd. for C29H48NO10, 570.3273).
Aconicarmichoside D (4): Colorless gum; [α]D20 +37.1 (c 0.50, MeOH); IR νmax 3360, 2937, 1678, 1445, 1359, 1310, 1201, 1126, 1041, 939, 899, 835, 801, 721 cm-1; 1H NMR (MeOH-d4, 600 MHz) data, see Table 1; 13C NMR (MeOH-d4, 150 MHz) data, see Table 1; (+)-ESIMS m/z 592 [M+Na]+, 570 [M+H]+; (-)-ESIMS m/z 604 [M +HCl-H]-, 568 [M-H]-; (+)-HR-ESIMS m/z 570.3277 [M+H]+ (calcd. for C29H48NO10, 570.3273).
4.4. Acid hydrolysis of 1-4Compounds 1-4 (1-2 mg) were separately dissolved in acetonitrile (0.1 mL), then refluxed with 2 mol/L HCl (2.0 mL) at 95 ℃ for 3 h. The reaction mixture was concentrated under reduced pressure and the residue was chromatographed by CC over Sephadex LH-20 (MeOH) to yield aglycone and sugar. The aglycone (0.5-1.2 mg) gave specific rotation{[α]D20 +20.7 to +22.3 (c 0.05-0.1, MeOH)} and 1H NMR spectroscopic data (MeOH-d4) consistent with those of neoline obtained from the same extract, while the sugar (0.1-0.4 mg) showed [α]D20 values of +104.8 to +110.1 (c 0.01-0.04, H2O) and 1H NMR spectroscopic data (D2O) are in good agreement with those of an authentic L-arabinose, which was also isolated from the plant extract.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (NNSFC, Nos. 81630094 and 30825044) is acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at 10.1016/j.cclet.2017.04.026.
| [1] | Jiangsu New Medical College, A Dictionary of Traditional Chinese Medicine, Shanghai Science and Technology Publishing House, Shanghai, 1995, pp. 228-2321191-1194. |
| [2] | Y. Chen, Y.L. Chu, J.H. Chu. Alkaloids of the Chinese drugs. Aconitum spp. IX. Alkaloids from Chuan-Wu and Fu-Tzu, Aconitum carmichaeli Debx, Acta Pharm. Sin. 12 (1965) 435–439. |
| [3] | J. Iwasa, S. Naruto. Alkaloids from Aconitum carmichaeli DEBX. J. Pharm. Soc. Jpn. 86 (1966) 585–590. DOI:10.1248/yakushi1947.86.7_585 |
| [4] | S.H. Shim, S.Y. Lee, J.S. Kim, K.H. Son, S.S. Kang. Norditerpenoid alkaloids and other components from the processed tubers of Aconitum carmichaeli. Arch. Pharm. Res. 28 (2005) 1239–1243. DOI:10.1007/BF02978206 |
| [5] | C. Konno, M. Shirasaka, H. Hikino. Structure of senbusine A, B and C, diterpenic alkaloids of Aconitum carmichaeli roots from China. J. Nat. Prod. 45 (1982) 128–133. DOI:10.1021/np50020a003 |
| [6] | W.D. Zhang, G.Y. Han, H.Q. Liang. Studies on the alkaloid constituents of Jiangyou Fu-zi Aconitum carmichaeli from Sichuan. Acta Pharm. Sin. 27 (1992) 670–673. |
| [7] | X.K. Wang, T.F. Zhao, S. Lai. A new N-formyl C19-diterpenoid alkaloids, aldohypaconitine, from cultivated Aconitum carmichaeli Debx. Chin. Chem. Lett. 5 (1994) 671–672. |
| [8] | J. Xiong, K. Gu, N.H. Tan. Diterpenoid alkaloids from the processed roots of Aconitum carmichaeli. Nat. Prod. Res. Dev. 20 (2008) 440–443465. |
| [9] | X.X. Liu, X.X. Jian, X.F. Cai, et al., Cardioactive C19-diterpenoid alkaloids from the lateral roots of Aconitum carmichaeli "Fu Zi". Chem. Pharm. Bull. 60 (2012) 144–149. DOI:10.1248/cpb.60.144 |
| [10] | F. Gao, Y.Y. Li, D. Wang, X. Huang, Q. Liu. Diterpenoid alkaloids from the Chinese traditional herbal "Fuzi" and their cytotoxic activity. Molecules 17 (2012) 5187–5194. DOI:10.3390/molecules17055187 |
| [11] | J. Zhang, G.B. Sun, Q.F. Lei, et al., Chemical constituents of lateral roots of Aconitum carmichaelii Debx. Acta Pharm. Sin. 49 (2014) 1150–1154. |
| [12] | G.H. Zhou, L.Y. Tang, X.D. Zhou, et al., A review on phytochemistry and pharmacological activities of the processed lateral root of Aconitum carmichaelii Debeaux. J. Ethnopharm. 160 (2015) 173–193. DOI:10.1016/j.jep.2014.11.043 |
| [13] | W.D. Xu, Y. Tian, Q.L. Guo, Y.C. Yang, J.G. Shi. Secoeuphoractin, a minor diterpenoid with a new skeleton from Euphorbia micractina. Chin. Chem. Lett. 25 (2014) 1531–1534. DOI:10.1016/j.cclet.2014.09.012 |
| [14] | Y. Tian, Q. Guo, W. Xu, et al., A minor Diterpenoid with a new 6/5/7/3 fused-ring skeleton from Euphorbia micractina. Org. Lett. 16 (2014) 3950–3953. DOI:10.1021/ol501760h |
| [15] | F. Wang, Y.P. Jiang, X.L. Wang, et al., Aromatic glycosides from the flower buds of Lonicera japonica. J. Asian Nat. Prod. Res. 15 (2013) 492–501. DOI:10.1080/10286020.2013.785531 |
| [16] | W.X. Song, Y.C. Yang, J.G. Shi. Two new β-hydroxy amino acid-coupled secoiridoids from the flower buds of Lonicera japonica:isolation, structure elucidation, semisynthesis, and biological activities. Chin. Chem. Lett. 25 (2014) 1215–1219. DOI:10.1016/j.cclet.2014.05.037 |
| [17] | Z.B. Jiang, W.X. Song, J.G. Shi. Two new 1-(6'-O-acyl-β-D-glucopyranosyl) pyridinium-3-carboxylates from the flower buds of Lonicera japonica. Chin. Chem. Lett. 26 (2015) 69–72. DOI:10.1016/j.cclet.2014.10.011 |
| [18] | Y. Yu, Z. Jiang, W. Song, et al., Glucosylated caffeoylquinic acid derivatives from the flower buds of Lonicera japonica. Acta Pharm. Sin. B 5 (2015) 210–214. DOI:10.1016/j.apsb.2015.01.012 |
| [19] | X. Wang, M. Chen, F. Wang, et al., Chemical constituents from root of Isatis indigotica. Chin. J. Chin. Mater. Med. 38 (2013) 1172–1182. |
| [20] | Y.F. Liu, M.H. Chen, X.L. Wang, et al., Antiviral enantiomers of a bisindole alkaloid with a new carbon skeleton from the roots of Isatis indigotica. Chin. Chem. Lett. 26 (2015) 931–936. DOI:10.1016/j.cclet.2015.05.052 |
| [21] | Y.F. Liu, M.H. Chen, Q.L. Guo, et al., Antiviral glycosidic bisindole alkaloids from the roots of Isatis indigotica. J. Asian Nat. Prod. Res. 17 (2015) 689–704. DOI:10.1080/10286020.2015.1055729 |
| [22] | Y.F. Liu, M.H. Chen, S. Lin, et al., Indole alkaloid glucosides from the roots of Isatis indigotica. J. Asian Nat. Prod. Res. 18 (2016) 1–12. DOI:10.1080/10286020.2015.1117452 |
| [23] | Y.F. Liu, X.L. Wang, M.H. Chen, et al., Three pairs of alkaloid enantiomers from Isatis indigotica. Acta Pharm. Sin. B 6 (2016) 141–147. DOI:10.1016/j.apsb.2016.01.003 |
| [24] | M.H. Chen, S. Lin, Y.N. Wang, et al., Antiviral stereoisomers of 3, 5-bis(2-hydroxybut-3-en-1-yl)-1, 2, 4-thiadiazole from the roots Isatis indigotica, Chin. Chem. Lett. 27(2016) 643-648. |
| [25] | D.W. Li, Q.L. Guo, X.H. Meng, et al., Two pairs of unusual scalemic enantiomers from Isatis indigotica leaves. Chin. Chem. Lett. 27 (2016) 1745–1750. DOI:10.1016/j.cclet.2016.08.006 |
| [26] | Y. Liu, M. Chen, Q. Guo, et al., Aromatic compounds from an aqueous extract of "ban lan gen" and their antiviral activities. ActaPharm. Sin. B 6 (2017) 179–184. |
| [27] | Y. Jiang, Y. Liu, Q. Guo, et al., Acetylenes and fatty acids from Codonopsis pilosula. Acta Pharm. Sin. B 5 (2015) 215–222. DOI:10.1016/j.apsb.2015.03.005 |
| [28] | Y.P. Jiang, Y.F. Liu, Q.L. Guo, et al., C14-polyacetylene glucosides from Codonopsis pilosula. J. Asian Nat. Prod. Res. 17 (2015) 601–614. DOI:10.1080/10286020.2015.1041932 |
| [29] | Y.P. Jiang, Q.L. Guo, Y.F. Liu, et al., Codonopiloneolignanin A, a polycyclic neolignan with a new carbon skeleton from the roots of Codonopsis pilosula. Chin. Chem. Lett. 26 (2016) 55–58. |
| [30] | Y. Jiang, Y. Liu, Q. Guo, et al., Sesquiterpene glycosides from the roots of Codonopsis pilosula. Acta Pharm. Sin. B 6 (2016) 46–54. DOI:10.1016/j.apsb.2015.09.007 |
| [31] | Q. Guo, Y. Wang, S. Lin, et al., 4-Hydroxybenzyl-substituted amino acid derivatives from Gastrodia elata. Acta Pharm. Sin. B 5 (2015) 350–357. DOI:10.1016/j.apsb.2015.02.002 |
| [32] | Q.L. Guo, Y.N. Wang, C.G. Zhu, et al., 4-Hydroxybenzyl-substituted glutathione derivatives from Gastrodia elata. J. Asian Nat. Prod. Res. 17 (2015) 439–454. DOI:10.1080/10286020.2015.1040000 |
| [33] | Q.L. Guo, S. Lin, Y.N. Wang, et al., Gastrolatathioneine, an unusual ergothioneine derivative from an aqueous extract of "tian ma":a natural product co-produced by plant and symbiotic fungus, Chin. Chem. Lett. (2016), doi:http://dx.doi.org/10.1016/j.cclet.2016.06.040. |
| [34] | B. Jiang, S. Lin, C.G. Zhu, et al., Diterpenoid alkaloids from the lateral root of Aconitum carmichaelii. J. Nat. Prod. 75 (2012) 1145–11591878. DOI:10.1021/np300225t |
| [35] | Z.B. Jiang, B.Y. Jiang, C.G. Zhu, et al., Aromatic acid derivatives from the lateral roots of Aconitum carmichaelii. J. Asian Nat. Prod. Res. 16 (2014) 891–900. DOI:10.1080/10286020.2014.939585 |
| [36] | Z.B. Jiang, X.H. Meng, B.Y. Jiang, et al., Two 2-(quinonylcarboxamino)benzoates from the lateral roots of Aconitum carmichaelii. Chin. Chem. Lett. 26 (2015) 653–656. DOI:10.1016/j.cclet.2015.04.011 |
| [37] | X.H. Meng, Z.B. Jiang, C.G. Zhu, et al., Napelline-type C20-diterpenoid alkaloid iminiums from "fu zi" and solvent/acid-dependent transformation/equilibration betweeniminium alcohol and aza-acetal forms. Chin. Chem. Lett. 27 (2016) 993–1003. DOI:10.1016/j.cclet.2016.05.013 |
| [38] | X.H. Meng, Z.B. Jiang, Q.L. Guo, J.G. Shi. A minor arcutine-type C20-diterpenoid alkaloid iminium constituent of "fu zi". Chin. Chem. Lett. 28 (2017) 588–592. DOI:10.1016/j.cclet.2016.11.010 |
| [39] | H. Hikino, Y. Kuroiwa, C. Konn. Structure of hokbusine A and B, diterpenic alkaloids of Aconitum carmichieli roots from Japan. J. Nat. Prod. 46 (1983) 178–182. DOI:10.1021/np50026a006 |
| [40] | J. Chen. Chemical constituent from radix Aconite Lateralis. Chin. Med. J. Res. Pract. 27 (2013) 33–35. |
| [41] | M. Halabalaki, A. Urbain, A. Parschali, et al., Quercetin and kaempferol 3-O-[α-L-rhamnopyranosyl-(1→2)-α-Larabinopyranoside]-7-O-α-L-rhamnopyranosides from Anthyllis hermanniae:structure determination and conformational studies. J. Nat. Prod. 74 (2011) 1939–1945. DOI:10.1021/np200444n |
| [42] | S.W. Pelletier, Z. Djarmati, S. Lajsic, W.H. De Camp. Alkaloids of Delphinium staphisagria. The structure and stereochemistry of delphisine, neoline, chasmanine, and homochasmanine. J. Am. Chem. Soc. 98 (1976) 2617–2625. DOI:10.1021/ja00425a035 |
| [43] | S.W. Pelletier, Z. Djarmati. Carbon-13 nuclear magnetic resonance:aconitinetype diterpenoid alkaloids from Aconitum and Delphinium species. J. Am. Chem. Soc. 98 (1976) 2626–2636. DOI:10.1021/ja00425a036 |
| [44] | W. Liu, X.J. Gou, Q. Song, F.Z. Chen, Neoline from Aconitum flavum Hand, Acta Cryst. E67(2011) o1435. |
| [45] | Z.T. Zhang, L. Wang, Q.F. Chen, et al., Revisions of the diterpenoid alkaloids reported in a JNP paper (2012, 75, 1145-1159). Tetrahedron 69 (2013) 5859–5866. DOI:10.1016/j.tet.2013.05.029 |
| [46] | F.P. Wang, D.L. Chen, H.Y. Deng, et al., Further revisions on the diterpenoid alkaloids reported in a JNP paper (2012, 75, 1145-1159). Tetrahedron 70 (2014) 2582–2590. DOI:10.1016/j.tet.2014.01.066 |
| [47] | C. Levrier, M.C. Sadowski, C.C. Nelson, R.A. Davis. Cytotoxic C20 diterpenoid alkaloids from the Australian endemic rainforest plant Anopterus macleayanus. J. Nat. Prod. 78 (2015) 2908–2916. DOI:10.1021/acs.jnatprod.5b00509 |
| [48] | J. Qu, L. Fang, X.D. Ren, et al., Bisindole alkaloids with neuralanti-inflammatory activity from Gelsemium elegans. J. Nat. Prod. 76 (2013) 2203–2209. DOI:10.1021/np4005536 |
| [49] | W.Y. He, R.M. Gao, X.Q. Li, J.D. Jiang, Y.H. Li. In vitro anti-influenza virus activity of 10 traditional Chinese medicines. Acta Pharm. Sin. 45 (2010) 395–398. |
| [50] | F.P. Wang, X.T. Liang, C20-diterpenoid alkaloids, in:G.A. Cordell (Ed.), The Alkaloids:Chemistryand Biology, Elsevier Science, New York, 2002, pp.1-280. |
| [51] | F.P. Wang, Q.H. Chen, The C19-diterpenoid alkaloids, in:G.A. Cordell (Ed.), The Alkaloids:Chemistry and Biology, Elsevier Science, New York, 2010, pp.1-577. |
| [52] | F.P. Wang, Q.H. Chen, X.T. Liang, The C18-diterpenoid alkaloids, in:G.A. Cordell (Ed.), The Alkaloids:Chemistry and Biology, Elsevier Science, New York, 2009, pp. 1-78. |
| [53] | F.P. Wang, Q.H. Chen, X.Y. Liu, Diterpenoid alkaloids, Nat. Prod. Rep. 27(2010) 529-570. |