 2017, Vol. 28
2017, Vol. 28 



b University of Chinese Academy of Sciences, Beijing 100049 China;
c Department of Hospital Pharmacy, University of Toyama, Toyama 930-0194, Japan;
d National Engineering Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, China
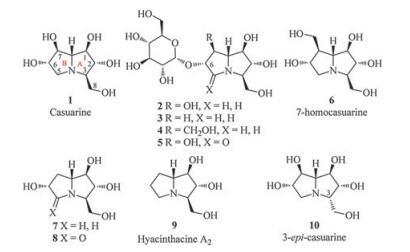
Casuarine (1) was first isolated from the bark of Casuarina equisetifolia L. (Casuarinaceae) in 1994 [1] and then from the leaves and bark of Eugenia uniflora L. (Myrtaceae) [2], which are traditionally used for treatment of cancer and diabetes, respectively. Casuarine (1) and its related analogues constitute an important class of the polyhydroxylated pyrrolizidines for their six continuous stereogenic centres and also the most-oxygenated bicylic framework. This class of alkaloids has been shown to exhibit attractive biological activities. For example, casuarine (1) was a potent inhibitor of processing glycosidase Ⅰ [3], rat intestinal maltase and rat intestinal isomaltase [4], and also showed potent inhibition of amyloglucosidase in a competitive manner [4, 5]. Importantly, casuarine (1) was able to inhibit human NtMGAM more strongly than acarbose [6]. The naturally occurring glucoside (2) not only retained the potent inhibition of amyloglucosidase of casuarine (1) [4, 5], but also was a more powerful trehalase inhibitor [4, 7] and notably one of the most potent inhibitors of Tre37 till now [6]. These noteworthy activities have endowed casuarine (1) and its derivatives promise for development of novel drugs against diabetes, cancer and HIV infection [6, 8], and therefore promoted the syntheses of a series of natural and non-natural structural analogues and subsequent structureactivity relationship (SAR) studies.
According to the report of Bonaccini et al. [5] for casuarine (1) and its analogues (2-9) (Fig. 1), the C-6 and C-7 positions have no significant influence on their inhibitory activity as amyloglucosidase and glucoamylase inhibitors, but the replenishment of a lactam structure at the C-5 positions (compounds 5 and 8) dramatically decreased their activities due to the block of ammonium cation formation and the limited conformational flexibility. Computational studies [5] showed that casuarine (1) interacted with the active site of A. niger amyloglucosidase through a hydrogen bond involving C-6 hydroxyl, whereas the C-7 hydroxyl did not present the correct geometry for hydrogen-bond formation. Therefore, the C-6 hydroxyl should be retained in potential glycosidase inhibitors, while the C-7 position could be modified by other electron donor, i.e., an amino group. In combination of our recent SAR study of polyhydroxylated pyrrolidines and pyrrolizidines [9] and the previous report of 3-epi-casuarine (10) for lost of most inhibitory activity [10], the relative configuration of A ring of casuarine (1) was thereof believed to be vital for efficient docking of the molecule with corresponding enzymes. Herein, we report the synthesis and glycosidase inhibition of casuarine derivatives with C-7 hydroxyl replaced by an aminomethyl or amide group.

|
Download:
|
| Fig. 1. Casuarine and related analogues. | |
{kind=link}
2. Results and discussions
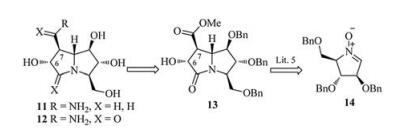
1, 3-Dipolar cycloaddition of polyhydroxylated cyclic nitrones with alkenes is amongst the most efficient methods of constructing the polyhydroxylated pyrrolizidine frameworks. Based on the previous efforts of Goti et al. [6, 11] and their cooperators [5], the target casuarine derivatives 11 and 12 could be achieved from the 1, 3-dipolar cycloaddition product 13 of nitrone 14 [12] and dimethyl maleate (Scheme 1).

|
Download:
|
| Scheme 1. Retrosynthesis of casuarine derivatives from nitrone. | |
{kind=link}
According to literature method [5], lactam 13 was obtained by the stereocontrolled cycloaddition of D-arabinose derived nitrone 14 followed by a subsequent reductive ring-opening/cyclization in 89% total yield. Lactam 13 was then put under ammonolysis procedure to give the expected amide 15a together with its C-7 epimer 15b in quantitative total yield (Scheme 2). The emergence of epimer 15b can be explained by the acidity of H-7 and the subsequent participation of an enol intermediate. The C-7 configuration of compound 15a was determined as S-configuration through NOESY experiments since a strong interaction of H-1 and H-7 was observed (See Supporting information), and the C-7 configuration of compound 15b was thereof determined as Rconfiguration. Since hydrogenolysis of 15a and 15b in existence of hydrochloric acid would lead to partial hydrolysis of the C-7 amides to the corresponding acids, the reaction was performed without addition of any acid, affording casuarine analougues 12 and 16 in excellent yield, respectively.

|
Download:
|
| Scheme 2. Synthesis of C-7 modified casuarine derivatives. | |
{kind=link}
While refluxing the lactams with LiAlH4 led to complex mixtures, reduction of 15a and 15b with NaBH4 in the presence of BF3·Et2O gave the corresponding amines 17a and 17b, respectively. 17b was obtained smoothly after purification by silica gel column chromatography, but purification of 17a was somewhat difficult due to the big polarity. 17a was thereof transformed to the N-Boc derivative 18, which can then be separated easily. Final hydrogenolysis of 18 and 17b according to routine procedures afforded the target casuarine analogues 11 and 19 respectively.
The synthesized C-7 derivatives of casuarines 11, 12, 16, 19 were assayed against a series of glycosidases (for details, see Supporting information). In contrast to casuarine (1) and 7-homocasuarine (6) [4, 5], lactam 12 and 16 are completely inactive to all the evaluated enzymes. Amine 11 was a weak inhibitor of yeast a-glucosidase (IC50 = 1000 mmol/L) and A. niger amyloglucosidase (IC50 = 151 mmol/L), and no significant inhibitory activities were observed for its C-7 epimer 19. The above assay results further confirmed the importance of reserving the free tertiary amine unit in effective interaction with enzymes. In the case of 11 and 19, though the replacement of C-7 hydroxymethyl with an aminomethyl group might not affect the capability of forming hydrogen bond with amino acid residues, addition of the primary amine group would obviously change pKa value of compounds 11 and 19, and finally affect their inhibitory activities.
3. ConclusionIn conclusion, casuarine analogues with C-7 aminomethyl or amide groups have been designed and synthesized from Darabinose-derived nitrones with 1, 3-dipolar cycloaddition as key step. Evaluation of the analogues against a series of glycosidases showed sharply decreased inhibitory activities, which may be attributed to obvious pKa change or limitation of configurations.
4. Experimental 4.1. General methodsAll reagents were used as received from commercial sources or prepared according to the literature. Analytical TLC were performed with 0.20 mm silica gel 60F plates and visualized by ultraviolet light or by treatment with a spray of Pancaldi reagent (NH4)6MoO4, Ce(SO4)2, H2SO4, H2O. Column chromatographic purification of products was carried out on silica gel (200– 300 mesh). Melting points were determined using an electrothermal melting point apparatus and were uncorrected. Infrared spectra were recorded on an FT-IR spectrometer. 1H NMR spectra were measured in CDCl3 (with TMS as an internal standard) or D2O on a magnetic resonance spectrometer (1H at 300 or 500 MHz, 13C at 75 MHz). High resolution mass spectra (HRMS) were recorded on an LTQ/FT linear ion trap mass spectrometer. Polarimetry was carried out using an Optical Activity AA-10R or Rudolph AutopolVI polarimeter and the measurements were made at the sodium Dline with a 0.5 dm or 1 dm path length cell. Concentrations (c) are given in gram per 100 mL.
4.2. 1, 2, 8-Tribenzyl-5-oxo-7-deoxy-7-methylcarboxylate casuarine (13)To a solution of nitrone 14 (7.0 g, 16.8 mmol) in dichloromethane (50 mL, distilled from CaH2) was added dimethyl maleate (4.2 mL, 33.6 mmol) and stirred overnight. After TLC showed completion of the reaction, the mixture was concentrated in vacuo, and the crude product was directly purified by flash column chromatography (silica gel, petroleum ether/EtOAc = 4:1, v/v) to give the cycloaddition product as a light yellow solid (9.2 g). The intermediate was then dissolved in acetic acid/water (55 mL, 10:1), followed by activated zinc powder (10.6 g, 0.16 mol), the suspension was stirred at 50 ℃ for 4 h until TLC showed completion of the reaction. The mixture was filtered, washed with EtOAc (3 × 50 mL), and then concentrated in vacuo. The residue was dissolved in dichloromethane/water (100 mL, 1:1), neutralized by aqueous NaHCO3 solution, and then extracted with dichloromethane (2 × 20 mL). The organic layers were combined, dried over MgSO4, and concentrated in vacuo. The crude product was recrystallized in petroleum ether/EtOAc (3:1, v/v) to give the target product 13 as a light yellow solid (7.9 g, 89% yield). Mp: 106–108 ℃; 1H NMR (300 MHz, CDCl3): δ 7.33–7.24 (m, 15H, PhCH2O), 4.77 (d, 1H, J = 9.6 Hz, H6), 4.56–4.43 (m, 6H, PhCH2O), 4.28–4.21 (m, 2H, H2 and H3), 3.98–3.89 (m, 2H, H1 and H7a), 3.77 (s, 3H, OMe), 3.58– 3.48 (m, 2H, H8), 3.04 (t, 1H, J = 9.0 Hz, H7); 13C NMR (75 MHz, CDCl3): δ 173.5, 171.4, 137.8, 137.43, 137.36, 128.50, 128.49, 128.4, 128.0, 127.9, 127.8, 127.7 (Ph), 87.4 (C1), 84.7 (C2), 74.3 (C6), 73.2, 72.2, 71.9 (PhCH2O), 68.2 (C8), 62.7 (C7a), 59.3 (C3), 54.8 (C7), 52.6 (Me) (Lit.[5]).
4.3. 1, 2, 8-Tribenzyl-5-oxo-7-deoxy-7-carboxamide casuarine (15a) and 1, 2, 8-tribenzyl-5-oxo-7-deoxy-7-epi-carboxamide casuarine (15b)To a suspension of compound 13 (3.61 g, 6.80 mmol) in methanol (50 mL) was added aqueous NH3 (15 mol/L, 50 mL), and stirred at room temperature for three days. After TLC showed completion of the reaction, the mixture was concentrated in vacuo, and purified by flash column chromatography (silica gel, MeOH/ EtOAc = 1:5, v/v) to afford the target product 15a (1.17 g, 33% yield) together with its C-7 epimer 15b (2.30 g, 66% yield), both as yellow solids.
Data for 15a: Mp: 111–113 ℃; [α]D20–21.1 (c 0.38 in MeOH); IR (KBr, cm-1): 3343 m, 2923 m, 2866 m, 1686 s, 1454 m, 1364 m, 1266 m, 1100 s, 1028 m, 736 s, 699 s; 1H NMR (500 MHz, CDCl3): δ 7.37–7.25 (m, 15H, PhCH2O), 6.30 (s, 1H, CONH2), 5.95 (s, 1H, CONH2), 4.95 (s, 1H, br, OH), 4.69 (dd, 2H, J = 26.5, 12.0 Hz), 4.59– 4.46 (m, 4H), 4.33–4.30 (m, 1H), 4.23 (t, 1H, J = 2.8 Hz), 4.14 (dd, 1H, J = 9.0, 5.0 Hz), 3.91 (t, 1H, J = 4.0 Hz), 3.56 (qd, 2H, J = 21.8, 6.5, 5.5 Hz), 2.90 (t, 1H, J = 10.0 Hz); 13C NMR (75 MHz, CDCl3): δ 174.0, 172.8, 137.9, 137.7, 137.6, 128.52, 128.48, 128.0, 127.9, 127.8 (Ph), 87.1, 85.1, 74.4, 73.2, 72.0, 71.9, 68.2, 61.6, 59.2, 54.9; HRMS (ESI) calcd. for C30H33N2O6+ 517.2333 [M + H]+, found 517.2337.
Data for 15b: Mp: 92–94 ℃; [α]D20 +15.0 (c 1.47 in MeOH); IR (KBr, cm-1): 3346 m, 2924 m, 1699 s, 1453 m, 1365 m, 1204 m, 1098 s, 1028 s, 737 m, 698 m; 1H NMR (300 MHz, CDCl3): δ 8.25 (s, br, 1H, ), 7.22–7.18 (m, 15H, PhCH2O), 4.85 (d, 1H, J = 9.4 Hz), 4.60– 4.35 (m, 6H, PhCH2O), 4.21–4.15 (m, 2H), 3.98–3.92 (m, 2H), 3.46 (s, 2H), 3.10 (t, 1H, J = 8.5 Hz); 13C NMR (75 MHz, CDCl3): δ 174.4, 174.1, 137.8, 137.60, 137.56, 128.5, 127.96, 127.92, 127.88, 127.82 (Ph), 87.4, 84.9, 74.6, 73.2, 72.3, 71.8 (PhCH2O), 68.1, 62.9, 59.3, 55.3; HRMS (ESI) calcd. for C30H33N2O6+ 517.2333 [M + H]+, found 517.2337.
4.4. 5-Oxo-7-deoxy-7-carboxamide casuarine (12) and 5-oxo-7-deoxy-7-epi-carboxamide casuarine (16)Compound 15a (0.30 g, 0.58 mmol) or 15b (0.21 g, 0.41 mmol) was dissolved in methanol (20 mL), followed by 10% Pd/C (30 mg). The suspension was stirred under hydrogen atmosphere for 48 h when TLC showed completion of the reaction. Hydrogen was replaced by nitrogen, catalyst was removed from the reaction mixture by filtration, and then washed with MeOH/H2O (1:1, 3 × 30 mL). The filtrate was concentrated in vacuo to give the target debenzylated product 12 (0.13 g, 91% yield) or 16 (0.09 g, 90% yield), both as light yellow syrup.
Data for 12: [α]D20–13.5 (c 0.74 in H2O); IR (KBr, cm-1): 3340 s, 2926 m, 1675 s, 1436 m, 1331 m, 1055 m; 1H NMR (300 MHz, D2O): δ 4.70 (s, 1H), 4.06 (dt, 1H, J = 6.6, 3.3 Hz), 3.81–3.77 (m, 1H), 3.76– 3.71 (m, 1H), 3.65 (t, 1H, J = 8.1 Hz), 3.59–3.54 (m, 2H), 2.96 (dd, 1H, J = 9.9, 8.1 Hz); 13C NMR (75 MHz, D2O): δ 174.2, 173.7, 79.6, 77.5, 74.4, 61.3, 61.1, 59.5, 55.3; HRMS (ESI) calcd. for C9H14N2O6Na+ 269.0744 [M + Na]+, found 269.0742.
Data for 16: [α]D20–17.8 (c 0.67 in H2O); IR (KBr, cm-1): 3340 s, 2919 m, 1694 s, 1422 m, 1275 m, 1193 m, 1032 m; 1H NMR (300 MHz, D2O): δ 4.74 (d, 1H, J = 10.2 Hz), 4.07 (t, 1H, J = 6.2 Hz), 3.83 (t, 1H, J = 7.2 Hz), 3.76–3.68 (m, 2H), 3.66–3.55 (m, 2H), 3.13–3.02 (m, 1H); 13C NMR (75 MHz, D2O): δ 173.7, 173.6, 79.7, 77.6, 73.9, 61.1, 59.7, 54.0; HRMS (ESI) calcd. for C9H14N2O6Na+ 269.0744 [M + Na]+, found 269.0742.
4.5. 1, 2, 8-Tribenzyl-7-deoxy-7-(tert-butoxycarbonyl)aminomethyl casuarine (18) and 1, 2, 8-tribenzyl-5-oxo-7-deoxy-7-epiaminomethyl casuarine (17b)To the solution of compound 15a or 15b (0.15 g, 0.29 mmol) in THF (20 mL, dried over 4 Å molecular sieve) was added NaBH4 (65.0 mg, 1.76 mmol) at 5 ℃. The mixture was stirred for 5 min, and BF3·Et2O (0.26 mL, 2.07 mmol) was added dropwise. The system was stirred at room temperature for 0.5 h, then heated at 50 ℃ for 3 h, TLC showed disappearance of the raw material. The suspension was cooled to room temperature, and poured into iced 1 mol/L HCl carefully, solvent and water was then removed in vacuo. The resulting mixture was basified by aqueous NaHCO3 solution, and extracted with EtOAc (3 × 20 mL). The organic layers were combined, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography (silica gel, MeOH/EtOAc = 1:10, v/v) to give the reductive product 17a (0.17 g, with impurities) or 17b (0.12 g, 85% yield), both as light yellow syrup. Compound 17a was then dissolved in methanol (20 mL), followed by K2CO3 (0.12 g, 0.87 mmol) and Boc2O (0.10 g, 0.55 mmol), and then reacted at room temperature for 1 h. After TLC showed completion of the reaction, solvent was removed in vacuo, and water (20 mL) as added. The mixture was extracted with EtOAc (3 × 20 mL), the organic phases were combined and dried over MgSO4, then concentrated in vacuo. The crude product were purified by flash column chromatography (silica gel, MeOH/ EtOAc = 1:10, v/v) to give the target product 18 (0.16 g, 94% yield for 2 steps) as a light yellow syrup.
Data for 18: [α]D20 +5.9 (c 3.07 in CH2Cl2); IR (KBr, cm-1): 3344 m, 2930 m, 1699 m, 1497 m, 1454 m, 1367 m, 1252 m, 1166 s, 1073 s, 1028 s, 738 m, 699 m; 1H NMR (300 MHz; CDCl3): δ 7.22– 7.12 (m, 15H, PhCH2O), 5.36 (s, 1H, NH), 4.51–4.39 (m, 8H, PhCH2O and OH), 4.05-3.97 (m, 3H), 3.57–3.31 (m, 5H), 3.14–3.09 (m, 1H), 3.02–2.94 (m, 1H), 2.88 (dd, 1H, J = 10.5, 5.4 Hz), 2.19 (t, 1H, J = 6.3 Hz), 1.50 (s, 2H, br, H2O), 1.30 (s, 9H, Boc); 13C NMR (75 MHz; CDCl3): δ 156.9, 137.8, 137.7, 137.6, 128.5, 128.4, 127.94, 127.89, 127.79 (Ph), 86.7, 84.6, 79.7, 74.7, 73.4, 72.6, 72.1, 70.8, 70.3, 69.3, 61.0, 51.8, 40.8, 28.4; HRMS (ESI) calcd. for C35H45N2O6+ 589.3272 [M + H]+, found 589.3272.
Data for 17b: [α]D20 +9.9 (c 1.62 in MeOH); IR (KBr, cm-1): 3347 m, 2870 m, 1454 m, 1366 m, 1074 s, 1028 s, 738 m, 698 m; 1H NMR (300 MHz, CDCl3): δ 7.32–7.19 (m, 15H, PhCH2O), 5.45 (s, br, 3H), 4.61–4.43 (m, 6H, PhCH2O), 4.32–4.28 (m, 1H), 4.16 (t, 1H, J = 4.5 Hz), 4.04 (t, 1H, J = 4.5 Hz), 3.73–3.49 (m, 7H), 3.08 (dd, 1H, J = 11.4, 4.8 Hz), 2.30 (t, 1H, J = 5.7 Hz); 13C NMR (75 MHz; CDCl3): δ 137.8, 137.6, 137.4, 128.5, 128.4, 128.0, 127.93, 127.89, 128.82, 127.77 (Ph), 86.6, 84.3, 74.1, 73.3, 72.6, 72.1, 70.4, 70.1, 69.0, 61.4, 61.3, 53.4; HRMS (ESI) calcd. for C30H37N2O4+ 489.2748 [M + H]+, found 489.2744.
4.6. 7-Deoxy-7-aminomethyl casuarine (11) and 5-oxo-7-deoxy-7-epi-aminomethyl casuarine (19)Compound 18 (0.06 g, 0.10 mmol) or 17b (0.08 g, 0.16 mmol) was dissolved in methanol (10 mL), followed by 10% Pd/C (30 mg) and 1 mol/L HCl (5 mL). The suspension was stirred under hydrogen atmosphere for 24 h when TLC showed completion of the reaction. Hydrogen was replaced by nitrogen, catalyst was removed from the reaction mixture by filtration, and then washed with MeOH/H2O for three times. The filtrate was concentrated in vacuo and neutralized with conc. NH3 and concentrated again. The residue was then purified by an acidic ion exchange column (Dowex 5W × 8-400, H+ form, Aldrich, column size: 1.3 cm × 14 cm), eluting with distilled water (100 mL) and then 1 mol/L NH4OH (50 mL), affording the target debenzylated product 11 (25.0 mg, 93% yield) or 19 (32.8 mg, 92% yield), both as light yellow syrup.
Data for 11: [α]D20 +32.5 (c 1.36 in MeOH); IR (KBr, cm-1): 3347 s, 2921 m, 1366 m, 1074 m, 1028 m; 1H NMR (300 MHz, D2O) δ 4.54 (dd, 1H, J = 10.8, 5.4 Hz), 4.41 (t, 1H, J = 7.4 Hz), 4.09 (dd, 1H, J = 9.0, 7.5 Hz), 3.99 (dd, 1H, J = 12.9, 3.3 Hz), 3.89 (dd, 1H, J = 5.4, 2.4 Hz), 3.85 (dd, 1H, J = 5.4, 1.5 Hz), 3.81 (d, 1H, J = 6.9 Hz), 3.77–3.71 (m, 1H), 3.46 (dd, 1H, J = 12.6, 5.7 Hz), 3.22 (d, 2H, J = 7.8 Hz), 2.85 (td, 1H, J = 13.5, 7.2 Hz); 13C NMR (75 MHz; D2O): δ 78.0, 74.5, 73.6, 72.3, 70.5, 59.0, 56.9, 47.2, 39.9; HRMS (ESI) calcd. for C9H19N2O4+ 219.1339 [M + H]+, found 219.1338.
Data for 19: [α]D20 +30.5 (c 1.64 in MeOH); IR (KBr, cm-1): 3344 s, 2925 m, 1368 m, 1095 m, 1032 m; 1H NMR (300 MHz; D2O): d 4.15 (d, 1H, J = 4.8 Hz), 4.04 (t, 1H, J = 7.8 Hz), 3.74–3.66 (m, 2H), 3.55–3.42 (m, 3H), 3.19 (dd, 1H, J = 11.4, 4.2 Hz), 3.03–2.95 (m, 2H), 2.81 (d, 1H, J = 7.1 Hz), 2.21 (t, 1H, J = 5.7 Hz); 13C NMR (75 MHz; D2O): δ 79.9, 77.0, 74.2, 70.6, 68.6, 61.9, 61.0, 60.2, 52.6; HRMS (ESI) calcd. for C9H19N2O4+ 219.1339 [M + H]+, found 220.1340.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (Nos. 21642012 and 21272240), National Science and Technology Major Projects for "Major New Drugs Innovation and Development" (No. 2013ZX09508104) and National Engineering Research Center for Carbohydrate Synthesis of Jiangxi Normal University are gratefully acknowledged. This work was supported in part by a Grant-in-Aid for Scientific Research (C) (No. 26460143) (AK) from the Japanese Society for the Promotion of Science (JSPS).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.06.016.
| [1] | R.J. Nash, P.I. Thomas, R.D. Waigh, et al., Casuarine:a very highly oxygenated pyrrolizidine alkaloid. Tetrahedron Lett 35 (1994) 7849–7852. DOI:10.1016/0040-4039(94)80134-7 |
| [2] | T. Matsumura, M. Kasai, T. Hayashi, et al., α-Glucosidase inhibitors from paraguayan natural medicine, Ñangapiry, the leaves of eugenia uniflora. Pharm. Biol 38 (2000) 302–307. DOI:10.1076/1388-0209(200009)3841-AFT302 |
| [3] | Y.T. Pan, H. Hori, R. Saul, et al., Castanospermine inhibits the processing of the oligosaccharide portion of the influenza viral hemagglutinin. Biochemistry 22 (1983) 3975–3984. DOI:10.1021/bi00285a038 |
| [4] | A. Kato, E. Kano, I. Adachi, et al., Australine and related alkaloids:easy structural confirmation by 13C NMR spectral data and biological activities. Tetrahedron:Asymmetry 14 (2003) 325–331. DOI:10.1016/S0957-4166(02)00799-1 |
| [5] | C. Bonaccini, M. Chioccioli, C. Parmeggiani, et al., Synthesis, biological evaluation and docking studies of casuarine analogues:effects of structural modifications at ring B on inhibitory activity towards glucoamylase, Eur.. J. Org. Chem (2010) 5574–5585. |
| [6] | F. Cardona, C. Parmeggiani, E. Faggi, et al., Total syntheses of casuarine and its 6-O-α-glucoside:complementary inhibition towards glycoside hydrolases of the GH31 and GH37 families. Chem. Eur. J 15 (2009) 1627–1636. DOI:10.1002/chem.v15:7 |
| [7] | M. Forcella, P. Parenti, L. Cipolla, et al., A membrane-bound trehalase from Chironomus riparius larvae:purification and sensitivity to inhibition. Glycobiology 20 (2010) 1186–1195. DOI:10.1093/glycob/cwq087 |
| [8] | (a) C. G. Bridges, S. P. Ahmed, M. S. Kang, et al. , The effect of oral treatment with 6-O-butanoyl castanospermine (MDL 28, 574) in the murine zosteriform model of HSV-1 infection, Glycobiology 5(1995) 249-253(b) R. J. Nash, PCT Int. Appl. WO2008009894 A2(2008). |
| [9] |
(a) Y. X. Li, M. H. Huang, Y. Yamashita, et al. , L-DMDP, L-homoDMDP and their C-3 fluorinated derivatives: synthesis and glycosidase-inhibition, Org. Biomol. Chem. 9(2011) 3405-3414; (b) Y. X. Li, Y. Shimada, K. Sato, et al. , Synthesis and glycosidase inhibition of australine and its fluorinated derivatives, Org. Lett. 17(2015) 716-719; (c) Y. Y. Song, K. Kinami, A. Kato, et al. , First total synthesis of (+)-broussonetine W: glycosidase inhibition of natural product & analogs, Org. Biomol. Chem. 14(2016) 5157-5174. |
| [10] | J.V. Ameijde, G. Horne, M.R. Wormald, et al., Isolation synthesis and glycosidase inhibition profile of 3-epi-casuarine. Tetrahedron:Asymmetry 17 (2006) 2702–2712. DOI:10.1016/j.tetasy.2006.10.005 |
| [11] | A. Brandi, F. Cardona, S. Cicchi, F.M. Cordero, A. Goti. Stereocontrolled cyclic nitrone cycloaddition strategy for the synthesis of pyrrolizidine and indolizidine alkaloids. Chem. Eur. J 15 (2009) 7808–7821. DOI:10.1002/chem.v15:32 |
| [12] |
(a) F. Cardona, E. Faggi, F. Liguori, M. Cacciarini, A. Goti, Total syntheses of hyacinthacine A2 and 7-deoxycasuarine by cycloaddition to a carbohydrate derived nitrone, Tetrahedron Lett. 44(2003) 2315-2318; (b) A. T. Carmona, R. H. Whightman, I. Robina, P. Vogel, Synthesis and glycosidase inhibitory activity of 7-deoxycasuarine, Helv. Chim. Acta 86(2003) 3066-3073; (c) S. Desvergnes, S. Py, Y. Vallée, Total synthesis of (+)-hyacinthacine A2 based on SmI2-induced nitrone umpolung, J. Org. Chem. 70(2005) 1459-1462; (d) W. B. Wang, M. H. Huang, Y. X. Li, et al. , A Practical synthesis of sugar-derived cyclic nitrones: powerful synthons for the synthesis of iminosugars, Synlett 3(2010) 488-492; (e) E. L. Tsou, Y. T. Yeh, P. H. Liang, W. C. Cheng, A convenient approach toward the synthesis of enantiopure isomers of DMDP and ADMDP, Tetrahedron 65(2009) 93-100. |