 2017, Vol. 28
2017, Vol. 28 



b School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China
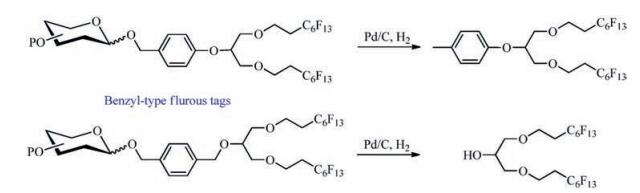
Carbohydrates, one of three major types of natural biomacromolecules, play important roles in life processes and are closely related to the development of many diseases [1]. Since the isolation of pure oligosaccharides from natural sources is extremely laborious and costly, synthesis of oligosaccharides is of importance for the acquisition of diverse oligosaccharides with well-defined structures. To improve the efficiency of oligosaccharide synthesis, many strategies for facilitating the purification process have been developed [2]. Among those, fluorous tagassisted purification by fluorous liquid–liquid extraction [3] (FLLE) or fluorous solid-phase extraction [4] (FSPE) have received considerable attentions in recent two decades [2h, 5]. However, the range of the application of FLLE in carbohydrate synthesis is limited due to the poor solubility of heavy fluorous tags (F content >60%) in general organic solvents and the high cost of fluorinated solvents used in the FLLE process. As an alternative to FLLE, FSPE has been developed rapidly in recent years with the use of light fluorous tags (F content < 40%) in the form of esters [5a, 5e, 6], silyl ethers [7], benzyl ethers [5c], acetals [8] and others [5b, 9], which are used as protecting groups to facilitate the purification of carbohydrate intermediates or in the syntheses of oligosaccharides [9e]. Especially, some light fluorous tags have been used in automated solution-phase synthesis of oligosaccharides [5b, 5f, 5g]. Nevertheless, these light fluorous tags more or less have some drawbacks, such as instability to acids or bases [9e], lower loading capacity [6b, 9e, 10], and difficulty in removal and regeneration. Therefore, it is necessary to develop a new type of fluorous tags with advantages of higher loading capacity than light fluorous tags and stability, good solubility in general organic solvent and ease to be removed and be regenerated.
Benzyl ethers, which are one of the most commonly used protecting groups and are stable to various reaction conditions in carbohydrate chemistry, are readily removable under catalytic hydrogenation conditions in the final stage of global deprotection of the synthetic oligosaccharides. Thus, we envisioned that benzyltype fluorous tags with double fluorous chains would be suitable for FSPE-assisted carbohydrates synthesis. The reasons for that are: (1) Tags with two fluorous chains could provide higher carbohydrate unit-loading capacity due to the higher fluorine content compared to those with one same fluorous chain [6b, 9e, 10]; (2) The benzyl-type fluorous tags might be cleaved together with other benzyl-type protecting groups from the target carbohydrate molecules by employing the same reaction conditions during the global de-benzylation, which thus avoid the extra process of removing fluorous tags (Scheme 1); (3) The fluorous tags could be easily regenerated in short steps with convenient operations. Herein, we'd like to report the preparation and application of the new designed recyclable benzyl-type fluorous tags.

|
Download:
|
| Scheme 1. Proposed benzyl-type fluorous tags with double chains and their removal from carbohydrates. | |
2. Results and discussion
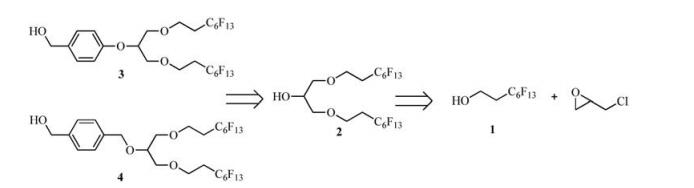
Retrosynthetically, the two benzyl-type fluorous tags (compounds 3 and 4) could be synthesized from the same key intermediate 2 (Scheme 2), which could be afforded via tandem Williamson etherification [11] and ring-opening of epoxide under base conditions with the use of commercially available starting material1, 1, 2, 2-tetrahydroperfluorooctanol 1 and epichlorohydrin.

|
Download:
|
| Scheme 2. Retrosynthetic analysis of tag 3 and 4. | |
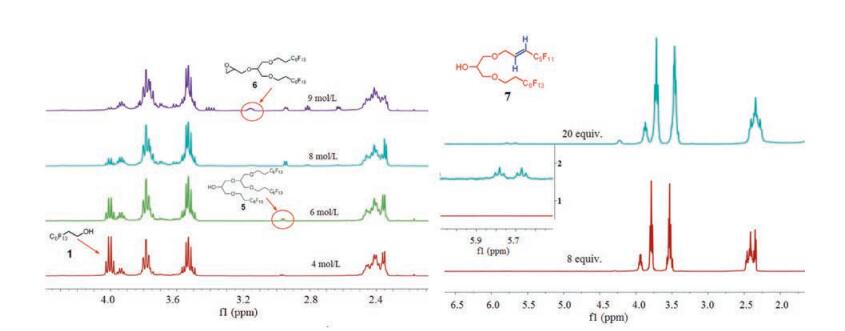
A series of studies revealed that aqueous NaOH is the appropriate base and that the amount of NaOH and the concentration of aqueous NaOH as well as the ratio of 1 to epichlorohydrin have great effects on the product formation. The results were summarized in Table 1, and the reactions were detected and analyzed by 1HNMR. The increase of the concentration of aqueous NaOH (Table 1, entries 1–4, 8 equiv. NaOH) could enhance the efficiency of the reaction. However, when the concentration was increased to 9mol/L, further Williamson etherification occurred between 2 and epichlorohydrin, leading to a side product, epoxide 6 (Fig. 1). Therefore, the optimal concentration was confirmed as 8mol/L. Subsequently, we discovered that with the increase of the amount of NaOH to 20 equiv., HF was eliminated from 2 to generate fluorous akene 7, which could not be separated from 2, but could be identified by 1HNMR (dt peakat the 5.7–5.8ppm, Fig. 1)and HRMS (ESI) (Table 1, entry 5). At last, 2 was obtained in 54% yield (entry 7) with a 1:2 ratio of alcohol 1 and epichlorohydrin. Furthermore, compound 5 (Fig. 1) which has similarly polarity with 2 was separated from the reaction mixture. As a heavy fluorous tag (the fluorous content is 62%), compound 5 could be used in synthesis of oligosaccharides assisted by FLLE.
|
|
Table 1 Investigation on the preparation of intermediate 2. |


|
Download:
|
| Fig. 1. 1H NMR under the different reaction conditions. | |
With compound 2 in hand, we next turned attention to synthesize compounds 3 and 4. To achieve the preparation of compound 3, Mitsunobu reaction [12] was applied to construct the skeleton of 3 (Scheme 3). Initially, the coupling reaction between 2 and 4-OH benzylaldehyde was carried out at 0 ℃ or room temperature in THF, and only a trace amount of the expected product was obtained. When the reaction temperature was increased to 100 ℃ in THF, the reaction proceeded smoothly and provided the product 8 in 48% yield. Under the similar condition, compound 8 could be afforded in 77% yield with the toluene as the alternative solvent. Reduction of 8 with NaBH4 yielded compound 3 quantitatively. To assemble compound 4, treatment of 2 with imidate 9 gave 10 in 90% yield in the presence of a catalytic amount of TMSOTf (0.1 equiv.) and 4Å MS at 0 ℃. Subsequent deprotection of the TBDPS group of 10 resulted in the formation of the target product 4 in 77% yield. Both tag 3 and 4 have good solubility in general organic solvents, such as CH2Cl2, MeCN, Et2O and toluene, which enable them tobe applicable toFSPE-assistedcarbohydrates synthesis. The fluorous tagged mono-or oligosaccharides were also soluble in DMF, THF, acetone, etc. In most cases, the F-tagged compounds can be purified from non-fluorous compound smoothly using common loading solvents for FSPE purification. However, if highly hydrophobic protecting groups (such as TBDPS) is attached to the F-tagged compounds, precipitation will occur in the presence of MeOH/H2O (as eluant to remove non-fluorous compounds) and thus leads to failure of the purification.

|
Download:
|
| Scheme 3. Syntheses of tags 3 and 4. | |
With success in the syntheses of tags 3 and 4, wenext examined the glycosylation of tag 3 and 4 with various imidate donors and the removal of the tags from tag-tethered carbohydrates. As indicated in Table 2, all of the imidate donors ("armed" or "disarmed") were coupled with both tags in good to high yield. Subsequently, cleavage of the tags from the corresponding coupling products was tested under catalytic hydrogenation conditions. The efficient deprotection occurred in a mixed solvent of MeOH/THF (1/3, v/v), rather than in MeOH. Thus, under the optimal conditions, the fluorous tags tethered to the coupling products were clearly removed to give the corresponding carbohydrates as well as 20 for tag 3-tethered carbohydrates or 2 for 4-tethered carbohydrates in excellent yield. Surprisingly, in entry 1, 33% of 20 was obtained along with the formation of 2 in 48% yield.
|
|
Table 2 Glycosylation of tag 3 and 4 and deprotection of tag-tethered carbohydrates. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
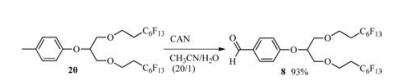
After accomplishing the deprotection of the fluorous tags from carbohydrates, the regeneration of both tags was examined. The oxidation of compound 20 by CAN via Nicolaou's procedure [13] provided compound 8 in excellent yield (Scheme 4), which was then directly reduced with NaBH4 to afford fluorous tag 3 in quantitative yield (Scheme 3). Obviously, compound 2, which was obtained from the deprotection of 4-tethered carbohydrates, can be reused as the key intermediate to synthesize the fluorous tag 3 or 4 via the aforementioned 2-step strategy (Scheme 3). Thus, the benzyl-type of fluorous tags can be readily removed and regenerated. According to the above results, tag 3 and 4 have similar characteristic in term of glycosylation and both can be removed via hydrogenation from tag-tethered carbohydrates, but they have difference in the strategies for their regeneration compared to tag 4, tag 3 could be more suitable for FSPE-assisted carbohydrate synthesis in terms of cost and time consumed.

|
Download:
|
| Scheme 4. Regeneration of compound 3. | |
{kind=link}
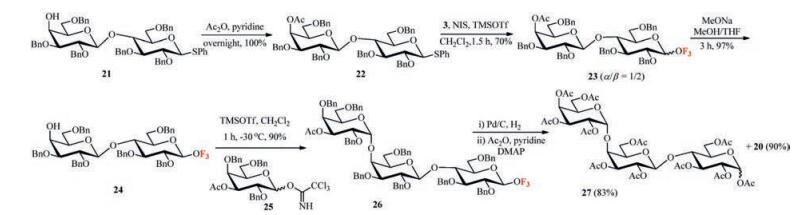
To demonstrate the utilities of the tags, oligosaccharides Gb3 was selected as the synthetic target. For this purpose, the compound 21 was prepared according to the reported methods [14] (Scheme 5). Acetylation of 21 with Ac2O gave 22 quantitatively. The reaction between 22 and 3 in the presence of NIS/ TMSOTf resulted in 23 (α/β, 1/2, determined by 1HNMR analysis of the mixture purified by FSPE) in 71% isolated yield. Treatment of the β anomer of 23 with MeONa/MeOH provided 24 in 97% yield, and then glycosylation of 24 with imidate 25 at -30 ℃ using TMSOTf as the promoter led to 26 in 90% yield. The global debenzylation of trisaccharide 26, however, resulted in complex mixtures under the catalytic hydrogenation condition with MeOH/ THF (1/3, v/v) as the solvent. Considering the addition of an acid is helpful to remove benzyl-type protecting groups, the solvent was changed to MeOH/AcOH/H2O (3/1/1, v/v/v) [15]. Gratifying, the fluorous tag and all of the benzyl protecting groups were cleaved together under 1.5 MPa hydrogen atmosphere. Nevertheless, HRMS (ESI) showed that the glycosidic linkage of the trisaccharide was broken under this condition, probably due to the acidic cleavage. Finally, the global debenzylation was achieved without cleavage of glycosidic bond by means of reducing the amount of AcOH to 4% [16]. Furthermore, treatment of the corresponding debenzylated product with Ac2O and pyridine gave the peracetylated Gb3 trisaccharide 27 in 83% yield along with the compound 20 in 90% yield over 2 steps.

|
Download:
|
| Scheme 5. Synthesis of per-acetylated Gb3. | |
{kind=link}
3. Conclusion
In conclusion, with cheaper starting materials, two benzyl-type fluorous tags were synthesized only in three steps. Introduction of the benzyl-type tags not only could simplify the separation process but also can serve as benzyl ether-type protective groups which are stable in general condition of coupling and deprotection in carbohydrate chemistry. This type of tags could be simultaneously removed together with normal benzyl protecting groups under catalytic hydrogenation conditions. In addition, both tags were regenerated in high yield via a 2-step procedure, especially the regeneration of tag 3 could be achieved efficiently with a low cost. Thus, it is expected that the process of FSPE-assisted oligosaccharide synthesis could become less time-consuming at a lower cost by using the benzyl-type tags. Furthermore, considering both tags possessing higher fluorine content (ca. 55%) than traditional light ones (F content < 40%), we expect the benzyl-type fluorous tags possess higher carbohydrate unit-loading capacity, and will open out a bright prospect in the FSPE-assisted synthesis of oligosaccharides with biological interests. Further application of the tags to the synthesis of complicated natural carbohydrates is in progress in our lab.
4. Experimental 4.1. Prepartion of fluorous tags1, 3-Bis[(3, 3, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 8-tridecafluorooctyl)oxy]-2-propanol (2): To a mixture of 1, 1, 2, 2-tetrahydroperfluorooctanol (2.00 g, 5.49 mmoL) and TBAI (0.61 mg, 1.65 mmoL) in a 50 mL flask was added aqueous NaOH (8 mol/L, 5.5 mL) and epichlorohydrin (0.65 mL, 8.23 mmoL). The mixture was stirred at 45 ℃ overnight, and then poured into H2O and extracted with Et2O, the organic layer was dried over MgSO4, concentrated and purified by silica gel chromatography (EtOAc/petroleum ether, 1/5, v/v) to give 2 (1.15 g, 1.47 mmol, 53%) and 5 (0.31 g, 0.26 mmol, 14%) both as clear oil. Compound 2: IR (KBr, cm-1): v 3310, 2890, 1482, 1404, 1362, 1241, 1141. 1HNMR (400 MHz, CDCl3): δ 4.03–3.84 (m, 1H), 3.87–3.71 (m, 4H), 3.60–3.45 (m, 4H), 2.50–2.30 (m, 5H, including 2.34 (d, 1H)). 13CNMR (100 MHz, CDCl3): δ 71.9, 69.2, 63.3 (t), 31.4 (t). HRMS (ESI) m/z Calcd. for C19H14F26O3Na: 807.0425 [M+Na]+, found: 807.0417. Compound 5: IR (KBr, cm-1): v 3433, 2921, 2882, 1384, 1237, 1132. 1HNMR (400 MHz, CDCl3): δ 4.10–3.76 (m, 5H), 3.76–3.58 (m, 6H), 3.58–3.23 (m, 6H), 2.94 (d, 1H, J = 4.2 Hz), 2.56–2.24 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 78.6, 72.0, 71.9, 71.0, 70.9, 69.6, 63.4–63.3 (m), 31.4 (t). HRMS (ESI) m/z Calcd. for C30H23F39O5Na: 1227.0820 [M+Na]+, found: 1227.0830.
4-(1, 3-Bis(3, 3, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 8-tridecafluorooctyloxy) propan-2-yloxy)-benzaldehyde (8): To a solution of compound 2 (0.73 g, 0.93 mmol), 4-hydroxybenzaldehyde (0.34 g, 2.82 mmol) and PPh3 (0.50 g, 1.91 mmol) in dry toluene (10 mL) was added DEAD (0.3 mL, 1.90 mmol) dropwise at 0 ℃. After the addition was complete, the mixture was heated to 110 ℃ and stirring for 2 h. The reaction mixture was concentrated in vacuo and purified by silica gel column chromatography (EtOAc/petroleum ether, 1/5-1/3, v/v) to afford compound 8 (0.63 g, 0.72 mmol, 77%) as a clear oil. IR (KBr, cm-1): v 2889, 2827, 1722, 1466, 1424, 1381, 1362, 1257, 1117, 1012. 1HNMR (400 MHz, CDCl3): δ 9.89 (s, 1H), 7.83 (d, 2H, J = 8.4 Hz), 7.07 (d, 2H, J = 8.3 Hz), 4.65 (p, 1H, J = 4.6 Hz), 3.85–3.76 (m, 4H), 3.75– 3.69 (m, 4H), 2.64–2.14 (m, 4H). 13CNMR (100 MHz, CDCl3): δ 190.6, 163.0, 132.0, 130.5, 115.9, 76.2, 69.8, 63.6 (t), 31.4 (t). HRMS (ESI) m/z Calcd. for C26H18F26O4Na: 911.0688 [M+Na]+, found: 911.0677.
4-(1, 3-Bis(3, 3, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 8-tridecafluorooctyloxy) propan-2-yloxy)benzenemethanol (3): To a solution of compound 8 (0.23 g, 0.26 mmol) in MeOH (3 mL) was added NaBH4 (12.8 mg, 0.34 mmol) at room temperature. After 1 h, NH4Cl (2 mL) was added, the resulting mixture was diluted with CH2Cl2, separated and the combined organic phase was dried over Na2SO4, filtered and concentrated in vacuo to give 3 (0.23 g, 0.26 mmol, 100%) as colourless oil. IR (KBr, cm-1): v 3395, 3066, 3038, 2883, 1610, 1509, 1424, 1361, 1245, 1008, 911, 737. 1HNMR (600 MHz, CDCl3): δ 7.29 (d, 1H, J = 8.6 Hz), 6.95 (d, 1H, J = 8.6 Hz), 4.62 (s, 1H), 4.49 (p, 1H, J = 4.9 Hz), 3.82–3.74 (m, 2H), 3.71 (qd, 2H, J = 10.2, 4.9 Hz), 2.40 (tt, 2H, J = 18.6, 6.6 Hz). 13CNMR (150 MHz, CDCl3): δ 157.5, 134.2, 128.7, 116.4, 76.5, 69.8, 64.91, 64.90, 63.5, 31.5 (t). HRMS (ESI) m/z Calcd. for C26H20F26O4Na: 913.0844 [M+Na]+, found: 913.0844.
(4-((1, 3-Bis(3, 3, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 8-tridecafluorooctyloxy) propan-2-yloxy)methyl)benzyloxy)(tert butyl)diphenylsilane (10): TMSOTf (23 mL TMSOTf in 1 mL CH2Cl2) was added to a solution of compound 2 (1.00 g, 1.27 mmol) in CH2Cl2 (5 mL) at 0 ℃, followed by a solution of compound 9 (1.98 g, 3.8 mmol) in CH2Cl2 (5 mL). After stirring for 1 h, Et3N (40 mL) was added, the mixture was purified by silica gel column chromatography (EtOAc/ petroleum ether, 1/5, v/v) to give compound 10 (1.30 g, 1.14 mmol, 90%) as a colorless syrup. IR (KBr, cm-1): v 3061, 2938, 2867, 1380, 1238, 704. 1HNMR (400 MHz, CDCl3): δ 7.75-7.65 (m, 4H), 7.44-7.34 (m, 6H), 7.32 (s, 4H), 4.76 (s, 2H), 4.66 (s, 2H), 3.81-3.71 (m, 4H), 3.71-3.67 (m, 1H), 3.62-3.54 (m, 4H), 2.49-2.26 (m, 4 H), 1.09 (s, 9H). 13CNMR (100 MHz, CDCl3): δ 140.7, 136.9, 135.6, 133.5, 129.7, 127.8, 127.7, 126.0, 76.6, 72.2, 70.8, 65.3, 63.3 (t), 31.5 (t), 26.8, 19.3. HRMS (ESI) m/z Calcd. for C43H40F26SiO4Na: 1165.2178 [M +Na]+, found: 1165.2198.
4-((1, 3-Bis(3, 3, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 8-tridecafluorooctyloxy) propan-2-yloxy)methyl)benzenemethanol (4): To a solution of compound 10 (1.29 g, 1.13 mmol) in dry THF (4 mL) was added TBAF (1 mol/L, 1.7 mL). After stirring at room temperature for 2 h, the solution was diluted with EtOAc, and then poured into H2O. The organic layer was separated, dried, and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc/ petroleum ether, 1/3, v/v) to give compound 4 (0.79 g, 0.87 mmol, 77%). IR (KBr, cm-1): v 2887, 1611, 1241, 1140, 705. 1HNMR (600 MHz, CDCl3): δ 7.34 (s, 4H), 4.69 (s, 2H), 4.66 (s, 2H), 3.79–3.71 (m, 4 H), 3.71–3.65 (m, 1 H), 3.63–3.50 (m, 4H), 2.30–2.47 (m, 4H); 13CNMR (150 MHz, CDCl3): δ 140.6, 137.8, 128.0, 127.0, 76.7, 72.0, 70.8, 64.9, 63.3 (t), 31.5 (t). HRMS (ESI) m/z Calcd. for C33H26F34O4Na: 1155.1288 [M + Na]+, found: 1155.1204.
4.2. Glycosylation of tag 3 and 4General procedure for glycosylation of fluorous 3/4 and imidate 11 using standard addition: To a solution of imidate 11 (2 equiv., 0.80 mmol) and 3/4 (0.40 mmol) in dry CH2Cl2 (2 mL) was added TMSOTf (10 mL TMSOTf was dissolved in 1 mL CH2Cl2, 0.75 mL) at -30 ℃. After stirring for 1 h, Et3N (20 mL) was added, the resulting mixture was directly purified by silica gel chromatography to give the product.
General procedure for glycosylation of fluorous 3/4 and imidate 12/13 using inverse addition: TMSOTf (10 mL TMSOTf was dissolved in 1 mL CH2Cl2, 0.75 mL) was added to a solution of 3/4 (0.40 mmol) in dry CH2Cl2 (2 mL) at -30 ℃. To the mixture was then added imidate 12/13 (2 equiv., 0.80 mmol). After 1 h, Et3N (20 mL) was added, the mixture was purified by silica gel chromatography to give the product.
4.3. Regeneration of compound 20To a solution of compound 20 (83 mg, 0.092 mmol) in CH3CN/ H2O (20/1, 3 mL) was added CAN (4 equiv.) in four portions during the course of 1 h. The resulting mixture was allowed to stir at room temperature for additional 1 h, and then poured into NaHCO3 aqueous and extracted with Et2O (3 × 10 mL). The organic layers was separated, dried and concentrated in vacuo, the residue was purified by silica gel column chromatography (EtOAc/petroleum ether, 1/5-1/3, v/v) to afford compound 8 (79 mg, 0.086 mmol, 93%).
4.4. Prepartion of oligosaccharides Gb3(4-(1, 3-Bis(3, 3, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 8-tridecafluorooctyloxy) propan-2-yloxy)phenyl)methyl O-(2, 3, 6-tri-O-benzyl-4-O-acetyl-β-D-galactopyronosyl)-(1→4)-2, 3, 6-tri-O-benzyl-β-D-glucopyronoside (23): To a mixture of compound 22 (1.37 g, 1.35 mmol), 3 (1.05 g, 1.18 mmol) and powdered 4 Å MS (2.50 g) in dry CH2Cl2 (9 mL) was added NIS (0.45 g, 2.00 mmol). After stirring at -30 ℃ for 10 min, a solution of TMSOTf in dry CH2Cl2 (32 mL in 1 mL CH2Cl2) was added dropwise. After 1 h, Et3N (60 mL) was added, the resulting mixture was filtered through Celite. The filtration was washed with Na2S2O3 (20 mL) and NaHCO3 aqueous (20 mL), the combined organic phase was dried over Na2SO4, concentrated and the residue was purified by FSPE cartridge (the ratio of α to β anomer was determined by 1H NMR analysis of the mixture purified by FSPE) and silica gel column chromatography (EtOAc/ petroleum ether, 1/5-1/3, v/v) to give β anomer (1.08 g, 0.60 mmol, 51%) and α anomer (0.41 g, 0.23 mmol, 19%) both as colorless syrup. β Anomer: 

(4-(1, 3-Bis(3, 3, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 8-tridecafluorooctyloxy) propan-2-yloxy)phenyl)methyl 2, 4, 6-tri-O-benzyl-3-O-acetyl-aglactopyranosyl-(1→4)-2, 3, 6-tri-O-benzyl-β-D-galactopyronosyl-(1→4)-2, 3, 6-tri-O-benzyl-β-D-glucopyronoside (26): To a solution of compound 24 (89 mg, 0.05 mmol) in CH2Cl2 (2 mL) was added TMSOTf (10 mL in 1 mL CH2Cl2, 0.1 mL) at -30 ℃. A solution of compound 25 (0.16 mol/L, 1 mL) in CH2Cl2 (1 mL) was added in two portions during 30 min. After stirring for additional 30 min, the reaction was quenched with Et3N (10 mL). The mixture was concentrated under reduced pressure and the residue was purified by FSPE cartridge to give a crude product (0.11 g, 0.05 mmol, 100%). The crude product was further purified by silica gel column chromatography (EtOAc/petroleum ether, v/v, 1/3) to give 26 (0.10 g, 0.04 mmol, 90%) as a colourless syrup. 
2, 3, 4, 6-Tetra-O-acetyl-α-D-galactopyranosyl-(1→4)-2, 3, 6-triO-acetyl-β-D-galactopyranosyl-(1→4)-1, 2, 3, 6-tetra-O-acetyl-D-glucopyranoside (Per-O-Ac globotriaose, 27): A solution of compound 26 (35 mg, 0.016 mmol) in EtOAc/MeOH/AcOH/H2O (2/3/0.24/0.16 mL) was kept stirring in the presence of 10% Pd/C (28.4 mg, 0.027 mmol) under 1.5 MPa hydrogen atmosphere for 4 days. The mixture was filtered through Celite and washed with MeOH, EtOAc and water. Concentrated, the residue was coevaporated with toluene to give a white solid. To the solid in a flask was added Ac2O (1 mL), pyridine (1 mL) and DMAP (6 mg), the mixture was stirred at 45 ℃ for 9 h and then stirred at 30 ℃ overnight, concentrated and purified by column chromatography (Ethyl acetate/petroleum, 3/1, v/v) on silica gel to give compound 27 (α/β=1/1, 13mg, 0.013mmol, 84%) as a white solid and fluorous tag 20 (13mg, 0.015mmol, 92%). 
The characterization data of compound 6, 14-20, 22, 24, spectra of NMR for all new compounds and known compounds are available in Supporting information.
4.5. General procedure for removing of fluorous tagA solution of a start material (0.1mmol) inTHF/MeOH (1/3) was stirring in the presence of 10% Pd/C (0.1mmol) under 1.5MPa hydrogen atmosphere for 3 days. The suspension was filtered through Celite, and then washed with EtOAc, MeOH and water. The combined filtrate was concentrated under reduced pressure. The residue was purified by FSPE to give the removed fluorous tag and oligosaccharides.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21072122, 21472119, 21302119) and Ministry of Science and Technology (No. 2012ZX09502-001-001). All of the financial supports are gratefully acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.06.020.
| [1] |
(a) C. R. Bertozzi, L. L. Kiessling, Chemical glycobiology, Science 291(2001) 2357-2364; (b) P. Sears, C. H. Wong, Toward automated synthesis of oligosaccharides and glycoproteins, Science 291(2001) 2344-2350; (c) P. H. Seeberger, Exploring life's sweet spot, Nature 437(2005) 1239; (d) P. H. Seeberger, Chemical glycobiology: why now? Nat. Chem. Biol. 5(2009) 368-372; (e) L. X. Wang, B. G. Davis, Realizing the promise of chemical glycobiology, Chem. Sci. 4(2001) 3381-3394. |
| [2] |
(a) Z. Zhang, I. R. Ollmann, X. S. Ye, et al. , Programmable one-potoligosaccharide synthesis, J. Am. Chem. Soc 121(1999) 734-753; (b) X. Huang, L. Huang, H. Wang, X. S. Ye, Iterative one-pot synthesis of oligosaccharides, Angew. Chem. Int. Ed. 43(2004) 5221-52244; (c) O. Kanie, Y. Ito, T. Ogawa, Orthogonal glycosylation strategy in oligosaccharide synthesis, J. Am. Chem. Soc. 116(1994) 12073-12074; (d) C. S. Bennett, Principles of modern solid-phase oligosaccharide synthesis, Org. Biomol. Chem. 12(2014) 1686-1698; (e) M. C. Galan, R. A. Jones, A. T. Tran, Recent developments of ionic liquids in oligosaccharide synthesis: the sweet side of ionic liquids, Carbohydr. Res. 375(2013) 35-46; (f) T. Nokami, R. Hayashi, Y. Saigusa, et al. , Automated solution-phase synthesis of oligosaccharides [1_TD$DIF]via iterative electrochemical assembly of thioglycosides, Org. Lett. 15(2013) 4520-4523; (g) J. Bauer, J. Rademann, Hydrophobically assisted switching phase synthesis: the flexible combination of solid-phase and solution-phase reactions employed for oligosaccharide preparation, J. Am. Chem. Soc. 127(2005) 7296-7297; (h) K. Goto, M. Mizuno, Application of fluorous chemistry for oligosaccharide synthesis, Trends Glycosci. Glycotechnol. 25(2013) 203-213 and references cited therein. |
| [3] | I.T. Horvath, J. Rabai. Facile catalyst separationwithout water:fluorousbiphase hydroformylation of olefins. Science 266 (1994) 72–75. DOI:10.1126/science.266.5182.72 |
| [4] |
(a) W. Zhang, Fluorous tagging strategy for solution-phase synthesis of small molecules, peptides and oligosaccharides, Curr. Opin. Drug Discov. Dev. 7(2004) 784-797; (b) W. Zhang, D. P. Curran, Synthetic applications of fluorous solid-phase extraction (F-SPE), Tetrahedron 62(2006) 11837-11865. |
| [5] |
(a) R. Roychoudhury, N. L. B. Pohl, Synthesis of fluorous photolabile aldehyde and carbamate and alkyl carbamate protecting groups for carbohydrateassociated amines, Org. Lett. 16(2014) 1156-1159; (b) S. L. Tang, N. L. B. Pohl, Automated solution-phase synthesis of β-1, 4-mannuronate and β-1, 4-mannan, Org. Lett. 17(2015) 2642-2645; (c) W. Huang, Q. Gao, G. J. Boons, Assembly of a complex branched oligosaccharide by combining fluorous-supported synthesis and stereoselective glycosylations using anomeric sulfonium ions, Chem. Eur. J. 21(2015) 12920-12926; (d) K. Fukuda, M. Tojino, K. Goto, et al. , A recyclable heavy fluorous tag carrying an allyl alcohol pendant group: design and evaluation toward applications in synthetic carbohydrate chemistry, Carbohydr. Res. 407(2015) 122-130; (e) C. Cai, D. M. Dickinson, L. Li, et al. , Fluorous-assisted chemoenzymatic synthesis of heparan sulfate oligosaccharides, Org. Lett. 16(2014) 2240-2243; (f) S. L. Tang, N. L. B. Pohl, Automated fluorous-assisted solution-phase synthesis of β-1, 2-, 1, 3-, and 1, 6-mannan oligomers, Carbohydr. Res. 430(2016) 8-15; (g) S. Bhaduri, N. L. B. Pohl, Fluorous-tag assisted syntheses of sulfated keratan sulfate oligosaccharide fragments, Org. Lett. 18(2016) 1414-1417. |
| [6] |
(a) L. Manzoni, R. Castelli, Froc: A new fluorous protective group for peptide and oligosaccharide synthesis, Org, Lett. 8(2006) 955-957; (b) G. Park, K. S. Ko, A. Zakharova, N. L. Pohl, Mono-vs. di-fluorous-tagged glucosamines for iterative oligosaccharide synthesis, J. Fluorine Chem. 129(2008) 978-982. |
| [7] |
(a) L. Manzoni, Rapid synthesis of oligosaccharides using an anomeric fluorous silyl protecting group, Chem. Commun. (2003) 2930-2931; (b) F. Zhang, W. Zhang, Y. Zhang, D. P. Curran, G. Liu, Synthesis and applications of a light-fluorous glycosyl donor, J. Org. Chem. 74(2009) 2594-2597. |
| [8] | M. Kojima, Y. Nakamura, S. Takeuchi. A practical fluorous benzylidene acetal protecting group for a quick synthesis of disaccharides. Tetrahedron Lett. 48 (2007) 4431–4436. DOI:10.1016/j.tetlet.2007.04.106 |
| [9] |
(a) H. Tanaka, Y. Tanimoto, T. Kawai, T. Takahashi, A fluorous-assisted synthesis of oligosaccharides using a phenyl ether linker as a safety-catch linker, Tetrahedron 67(2011) 10011-10016; (b) C. Zong, A. Venot, O. Dhamale, G. J. Boons, Fluorous supported modular synthesis of heparan sulfate oligosaccharides, Org. Lett. 15(2013) 342-345; (c) B. Yang, Y. Jing, X. Huang, Fluorous-assisted one-pot oligosaccharide synthesis, Eur. J. Org. Chem. (2010) 1290-1298; (d) Y. Jing, X. Huang, Fluorous thiols in oligosaccharide synthesis, Tetrahedron Lett. 45(2004) 4615-4618; (e) R. Roychoudhury, N. L. B. Pohl, Light Fluorous-Tag-Assisted Synthesis of Oligosaccharides, in: D. B. Werz, S. Vidal (Eds. ), Mod. Synth. Methods Carbohydr. Chem. , Wiley-VCH, Weinheim, 2014, pp. 221-239 and references cited therein. |
| [10] | T. Miura, K. Goto, D. Hosaka, T. Inazu. Oligosaccharide synthesis on a fluorous support. Angew. Chem. Int. Ed. 42 (2003) 2047–2051. DOI:10.1002/anie.200250531 |
| [11] |
(a) B. Boutevin, B. Youssef, S. Boileau, A. M. Garnault, Synthese d'ethers et de thioethers allyliques fluores par catalyse par transfert de phase, J. Fluorine Chem. 35(1987) 399-410; (b) S. Malfait, S. Gérard, R. Plantier-Royon, G. Mignani, C. Portella, Synthesis of bi-and tetracatenar highly fluorinated compounds for grafting on silicone materials, J. Fluorine Chem. 132(2011) 760-766; (c) W. J. Huang, C. Y. Jin, D. K. Derzon, et al. , Synthesis of ether-linked fluorocarbon surfactants and their aggregational properties in organic solvents, J. Colloid Interface Sci 272(2004) 457-464. |
| [12] | M. Mizuno, S. Kitazawa, K. Goto. p-Alkoxyphenyl-type heavy fluorous tag for the preparation of carbohydrate units. J. Fluorine Chem. 129 (2008) 955–960. DOI:10.1016/j.jfluchem.2008.06.013 |
| [13] | K.C. Nicolaou, D. Sarlah, T.R. Wu, W. Zhan. Total synthesis of hirsutellone B. Angew. Chem. Int. Ed. 48 (2009) 6870–6874. DOI:10.1002/anie.v48:37 |
| [14] | B. Sun, A.V. Pukin, G.M. Visser, H. Zuilhof. An efficient glycosylationreactionfor the synthesis of asialo GM2 analogues. Tetrahedron Lett. 47 (2006) 7371–7374. DOI:10.1016/j.tetlet.2006.08.008 |
| [15] | Z. Wang, L. Zhou, K. El-Boubbou, X.S. Ye, X. Huang. Multi-component one-pot synthesis of the tumor-associated carbohydrate antigen Globo-H based on preactivation of thioglycosyl donors. J. Org. Chem. 72 (2007) 6409–6420. DOI:10.1021/jo070585g |
| [16] | A. Chernyak, S. Oscarson, D. Turek. Synthesis of the Lewisb hexasaccharide and squarate acid-HSA conjugates thereof with various saccharide loadings. Carbohydr. Res. 329 (2000) 309–316. DOI:10.1016/S0008-6215(00)00189-0 |
| [17] |
(a) M. R. E. Aly, P. Rochaix, M. Amessou, L. Johannes, J. C. Florent, Synthesis of globo-and isoglobotriosides bearing a cinnamoylphenyl tag as novel electrophilic thiol-specific carbohydrate reagents, Carbohydr. Res. 341(2006) 2026-2036; (b) H. W. Hsieh, M. W. Schombs, J. Gervay-Hague, Integrating ReSET with glycosyl iodide glycosylation in step-economy syntheses of tumor-associated carbohydrate antigens and immunogenic glycolipids, J. Org. Chem. 79(2014) 1736-1748. |