 2017, Vol. 28
2017, Vol. 28 



b Marine Biomedical Research Institute, Qingdao 266071, China
Dysfunctional protein-protein interactions (PPIs) is often associated with many diseases including cancer, diabetes and autoimmune disorders. Consequently, molecules that selectively target and/or inhibit PPIs exhibit great therapeutic potential and attract much attention. The successful PPIs inhibitors require the ability to mimicking the size and structure of protein interaction domains and the structure of these molecules should be flexible enough to convert to an induced active binding conformation [1, 2]. Many PPIs with large protein contact surfaces are difficult to target with small molecules [3, 4]. In contrast, natural or modified peptides with proper-length sequence can target these PPIs with high affinity and selectivity to the large protein contact surfaces. However, the rapid degradation by proteases or proteolytic enzymes and poor bioavailability has severely limited the development of peptide-based therapeutics [5]. Therefore, peptide-based inhibitors of PPIs require high affinity, selectivity and bioavailability to be effective both in vitro and in vivo.
Bcl-2 family proteins composed of pro-and anti-apoptotic members and served as essential regulators of the balance between cell life and death through the mitochondrial apoptotic pathway via interactions among different members [6, 7]. Two types of this family proteins promote apoptosis: the BH3-only (because they have only the BH3 domain of homology to Bcl-2) proteins (such as Bim, Bid and Bad) sense cellular stresses/damages and activate Bax and Bak either directly or indirectly through inhibition of the anti-apoptotic proteins, and the activated proapoptotic proteins Bax and Bak trigger apoptosis by permeabilizing the outer mitochondrial membrane, releasing apoptogenic factors such as cytochrome c that promote activation of caspases (proteases that mediate cellular demolition). The other type of Bcl-2 proteins, anti-apoptotic members (for example, Bcl-2, Bcl-xL and Mcl-1) promote cell survival by opposing the activation of Bax and Bak. Whereas, these guardians become inactivated when the BH3 domain (an α-helix) of BH3-only proteins binds to the hydrophobic groove formed by multi-domains of the antiapoptotic proteins. Consequently, inhibition of anti-apoptotic Bcl-2 proteins and/or up-regulation of BH3-only proteins enhance apoptosis [8-13]. Accordingly, the anti-apoptotic Bcl-2 proteins have been regarded as a promising target to develop anti-cancer drugs due to their overexpression in many cancers. One of the strategies is termed as 'BH3 mimetics': molecules that can mimic the α-helical BH3 domain of BH3-only proteins via binding to the shallow, hydrophobic cleft on the surface of the target Bcl-2 proteins with high affinity, inducing Bax/Bak activation and apoptosis in cancer cells [13-15]. Structural analysis of crystal complexes of BH3 domains and anti-apoptotic family members has revealed that BH3 domains featured four hydrophobic residues in an i, i + 4, i + 7, i + 11 pattern, which aligned along one face of the helix and occupied pockets within the BH3-recognition cleft of anti-apoptotic Bcl-2 proteins [9, 16, 17]. The 'BH3 mimetics' strategy has great potential in the development of inhibitors to treat Bcl-2 mediated diseases such as cancer. And the candidate inhibitors may also serve as useful tools to explore novel biological functions of the Bcl-2 family proteins [15, 18]. However, the development of peptide-based anticancer drugs targeting anti-apoptotic Bcl-2 proteins still remains as a tremendous challenge. In the past decades, there have been significant advancement in the development of peptide-based BH3 domain mimetics targeting the interaction between BH3-only proteins and anti-apoptotic Bcl-2 proteins. Notably, some groups have developed peptide mimics of segment of natural BH3 domain with high binding affinity and/ or antitumor effect in cell lines and animal models via introducing site-mutated, side chain cross-linked, α/β-and/or hydrocarbonstapled peptide fragments to stabilize binding conformation (an α-helix) with the hydrophobic cleft [19-27]. In vitro bioassay results indicated that these peptide BH3 mimetics were attractive candidates for inhibiting or disrupting BH3: Bcl-2 interactions, however, their efficacy in vivo was severely reduced due to the loss of secondary structure and/or low biostability/bioavailability.
Besides the above strategies to stabilize the α-helix, we have adopted an alternative strategy in which we postulated that the homology portion of BH3 domains containing the four conserved hydrophobic residues (Scheme 1, h1-h4) was the minimum sequence required for the bioactivity and the "active core" region. Recent studies have illustrated that the shortened GLP-1 (glucagon-like peptide-1) analogues reserving the homology portions but deleting the heterogenic portions showed superior bloodglucose-lowering effect to that of GLP-1 [28]. Herein, we synthesized seven analogues of Bim BH3 domain by truncating or modifying the C-and N-terminal of the lead Bim peptide (Scheme 1) and tested their binding affinity to anti-apoptotic Bcl-2 proteins and anticancer effects against different cancer cell lines.

|
Download:
|
| Scheme1. Design strategy for the Bim BH3 domain analogues targeting antiapoptotic Bcl-2 proteins | |
2. Results and discussion 2.1. Design of Bim-derived analogues
BH3 domains of pro-apoptotic BH3-only proteins all contain four conserved hydrophobic residues (Scheme 1, h1-h4). These four side chains along one face of the BH3 α-helix interacted with anti-apoptotic Bcl-2 proteins by occupying pockets within the BH3-recognition cleft. The BH3 domain peptide derived from Bim was found to bind tightly to all five anti-apoptotic proteins, which make it multi-target potency. Previous studies indicated that Bak 11-mer, a 11-residue peptide containing hydrophobic residue h2, h3 and h4 derived from Bak exhibited no interaction with Bcl-xL, whereas, the 16-mer peptide containing all the four hydrophobic residues showed tight binding affinity [9]. Interestingly, the 16-mer peptide derived from the same region of Bad exhibited little or no interaction with Bcl-xL. The length extended to 26 amino acids peptides derived from Bad were found to bind tightly to Bcl-xL with a KD of 6.0 nmol/L [29]. These results suggested the length of the peptide can significantly influence the binding affinity. Therefore, before designing the Bim-derived analogues, we examined the effect of Bim peptide truncation on binding affinity, and found the 18-mer and 21-mer Bim-derived peptides all proved to be comparable to the parental 26-mer peptide in terms of binding affinity [22-25]. Upon the previous finding that the sequencesimplified GLP-1 analogue only containing the functional homology portion exhibited prolonged blood-glucose-lowering effects [28], we proposed a hypothesis that the 12-mer Bim-derived peptide containing the four critical hydrophobic side chains might be the minimum sequence required for bioactivity and the "active core" region. To verify the hypothesis, 7 analogues were designed and synthesized via modification/simplification of the N-and/or Cterminal of the 21-mer Bim lead peptide (Scheme 1).
2.2. Structural analysis in solution by circular dichroismTo gain insight into the impact of the modification/simplification on analogues conformation in solution, we determined the far-UV circular dichroism (CD) spectra of Bim-derived analogues containing the hypothesized active core (Fig. 1). Consistent with previous report [24], the lead Bim peptide showed few evidence of helicity in buffer solution. Whereas, the hypothesized core region modified analogue SM-6 showed an a-helical-like signature in the far-UV CD, with minimum at 220 nm and characteristic absorption at 190nm, 208 nm. Interestingly, other analogues including sequence-truncated analogues SM-1-SM-5 and hypothesized core region modified analogue SM-7 showed as few evidence of a-helical signature as the lead Bim did. These results indicate that sequence-truncation of the lead Bim alone did not exert significant effect on the helical conformation. Nevertheless, we propose hypothesis that modification of the suitable fragments or groups within the hypothesized core region might affect the helix stability.

|
Download:
|
| Fig. 1. Far-UV CD spectra of the analogues at 10 mmol/L in solution (50 mmol/L Tris, 100 mmol/L NaCl, pH 8.0) at 20 ℃ | |
2.3. Protein binding affinity by SPR
Direct binding affinity of the Bim-derived analogues to the two key anti-apoptotic proteins, Bcl-2 and Bcl-xL were measured using the surface plasmon resonance (SPR) technique. The results in Table 1 showed that the lead Bim peptide, SM-1, SM-5 and SM-6 bound to both Bcl-2 and Bcl-xL, whereas no specific binding affinity of SM-3 were detected to either Bcl-2 or Bcl-xL. SM-2, SM-4 and SM-7 were found to bind to only one of the anti-apoptotic proteins. SM-1, SM-2, SM-4 and SM-6 all bound to Bcl-2 as tightly as the lead Bim peptide. Whereas, SM-5, the hypothesized active core, showed stronger binding affinity to Bcl-2 than the lead Bim. Specifically, the helical analogue SM-6 exhibited declined binding affinity compared with the lead Bim (~5-fold declined) and SM-5 (~8-fold declined). SM-1 and SM-2 both retained similar binding affinity to Bcl-xL with the lead Bim, and SM-5 bound to Bcl-xL with significantly improved affinity (~280-fold). However, SM-7 only retained ~50-fold declined binding affinity for Bcl-xL. Interestingly, the 12-mer SM-5, the shortest sequence containing the four critical hydrophobic side chains retained relatively high affinity for Bcl-2 but displayed reduced affinity for Bcl-xL, as compared with the parent Bim peptide. On the contrary, SM-6 derived from modifying the N-terminal of 12-mer sequence exhibited significantly high affinity for Bcl-xL but reduced affinity for Bcl-2. The other homologue SM-7 showed specific binding affinity to neither Bcl-2 nor Bcl-xL.
|
|
Table 1 Binding affinity to anti-apoptotic proteins determined by SPR |

{kind=link}
{kind=link}
Taken together, these results indicate that the sequencetruncation of lead Bim does not negatively impact the affinity with the anti-apoptotic proteins. Indeed, the SM-5:Bcl-2 and SM-6: Bcl-xL interactions are stronger than the lead Bim.
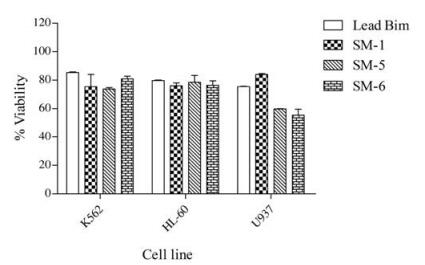
2.4. Cell killing activityPrevious studies have shown that stapled BH3 domain peptides can selectively induce apoptosis in some cell types, particularly hematological cancer cell lines [21, 24, 30-32]. We therefore determined the cell killing activity of these Bim BH3 analogs in three hematological cancer cell lines, HL-60, K562 and U937 using MTT assays. The results shown in Fig. 2 indicated that Bim BH3 mimetics exert relatively higher cell killing activity in U937 lymphoma cells than in HL-60 and K562 cells, especially SM-5 and SM-6 exhibited suppressing effect on cell viability in U937 lymphoma.

|
Download:
|
| Fig. 2. Viability of three cell lines (relative to DMSO control) when exposed to 50mmol/L analogues for 48h, as measured using MTTassay. Each bar represents the mean ±SD for three independent experiments | |
{kind=link}
However, the cell-killing activity of these Bim BH3 analogs are less potent as expected, although the binding assays showed that SM-5 and SM-6 could bind tightly to Bcl-2 or Bcl-xL (Table 1). One factor for the lack of cellular activity might be due to the intrinsic hydrophilicity of analogues which made them hard to entry cells and remain trapped within intracellular vesicles. The second explanation might be that analogues were degraded before reaching the cytoplasm of cells by proteases or proteolytic enzymes, which is a common defect of peptide-based potential therapeutic agents. The third possibility might be that the loss or destruction of α-helical secondary structure of analogues in cytoplasm led to the declined binding affinity to the target antiapoptotic proteins.
3. ConclusionIn summary, we have described the synthesis and bioactivity of seven analogues targeting anti-apoptotic Bcl-2 proteins derived from the Bim BH3 domain via sequence simplification and/or modification. This study demonstrated that the 12-mer analogue (SM-5) sequence containing the four hydrophobic side chains occupying pockets within the BH3-recognition cleft of antiapoptotic Bcl-2 proteins might be the minimum region required for bioactivity and the active core of Bim. Promising BH3 peptide mimetics with high binding affinity towards anti-apoptotic Bcl-2 proteins and significant cell-killing activity can be designed based on this "active core". Ultimately, analogues design based on the active core might serve as starting points for the development of potential anticancer/therapeutic agents.
4. Experimental 4.1. MaterialsRink Amide-AM resin (0.56 mmol/g), O-benzotriazole-N, N, N', N'-tertramethyl uronium hexafluorophosphate (HOBt), 1-hydroxybenzotriazole anhydrate (HBTU), N, N'-diisopropylethylamine (DIEA), N, N'-dissoprophlcarbodiimide (Dic), trifluoroacetic acid (TFA), Fmoc-protected amino acids were purchased from GL Biochem, Ltd. (Shanghai, China). High-performance liquid chromatography (HPLC)-grade N, N-dimethylformamide (DMF), dichloromethane (DCM) and methanol were purchased from Adamas-beta Co., Ltd. (Jiachuan Road, Shanghai, China). Acetic anhydride, hexadecanoic acid and polyethylene glycol methyl acid were purchased from Alfa Aesar, USA. Unless stated, reagents and solvents were not dried or further purified.
4.2. Analogues synthesis and purificationAll the analogues were synthesized manually on Rink AmideAM resin using the standard Fmoc strategy SPPS (solid phase peptide synthesis) with HOBt/HBTU/DIEA activation and piperidine Fmoc deprotection in a glass reaction vessel fitted, as previously described [33]. Full experimental details can be found in the Supporting information (SI). Crude peptides were cleaved from the resin and purified using the C18 reversed-phase semipreparative HPLC. After purification, analogue purity was assessed by analytical reverse-phase HPLC and product masses were confirmed by MALDI-TOF-MS. (See Table S1 and Fig. S1 in Supporting information).
4.3. Circular dichroism (CD)To evaluate the helical propensity of the Bim BH3 domain analogues, the far-UV circular dichroism (CD) spectra of each peptide which were dissolved at a final concentration of 10 mM in aqueous binding buffer (50 mmol/L Tris, 100 mmol/L NaCl, pH 8.0) were determined and analyzed by wavelength dependent circular dichroism (CD) spectroscopy (Fig. 2). Wavelength scans were performed on a Chirascan spectropolarimeter (Applied Photophysics Limited, UK) at 20 ℃. Each spectrum represents an average of five scans (background subtracted). CD measurements were collected as mean residue ellipticity (MRE) from samples at 20 ℃ spanning a wavelength range from 195 nm to 245 nm with a wavelength step size of 1 nm and a 10 s averaging time.
4.4. Recombinant protein expression and purificationHuman anti-apoptotic proteins Bcl-2 and Bcl-xL were expressed in Escherichia coli with a modified pET-28a vector encoding Histags. Briefly, Escherichia coli were harvested and lysed after induction by 1 mmol/L isopropy-β-D-thiogalactoside (IPTG). The supernatant was applied to the Ni-IDA sefinose resign purchased from Sangon Biotech (Shanghai) Co., Ltd., purified with elution buffer (50 mmol/L Tris, 500 mmol/L NaCl, pH 8.0) containing a gradient concentration of imidazole (0-200 mmol/L). To determine overall purity of the eluted protein, samples (20 mL) of the collected fractions were loaded onto a 10% polyacrylamide gel and separated by SDS-PAGE (Full details please see Supporting information text and Fig. S2).
4.5. Binding affinity measurementsDirect affinity measurements for binding of the analogues to Bcl-2 and Bcl-xL were performed using the Plexera PlexArray HT system (Plexera LLC, Woodinville, WA, USA) as described previously [34]. Briefly, peptides were immobilized to the surface of a biochip via UV crosslinking under the UV light (365 nm, 15 min) using the SpotBot3 Microarrayer system. Recombinant His-tagged Bcl-2 or Bcl-xL proteins (1 mg/mL in PBST buffer) was used and the interactions between the peptides with different Bcl-2 proteins were determined by SPR imaging using the Plexera PlexArray HT system.
4.6. Cell viability assayCell viability was evaluated using MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay, which is based on the ability of viable cells to convert water-soluble yellow tetrazolium salt into water-insoluble purple formazan product. The enzymatic reduction of the tetrazolium salt occurs only in living, metabolically active cells, but not in dead cells. Briefly, cells were seeded in 96-well plates at a density of 5 ×104 cells per well and subsequently treated with different BH3 peptides (50 mmol/L) at 37 ℃ for 48 h. Ten microliters of MTT labeling reagent was then added to each well and were incubated at 37 ℃ for 4 h. The precipitate was solubilized overnight and then quantitated by an ELISA microplate reader.
AcknowledgmentsThis study was financially supported by Postdoctoral Applied Research Project of Qingdao (No. 861605040085, to CZ, SW) and Grant of Innovation Plan in Biomedical Research of Qingdao City (No. 15-10-3-15-(28)-zch, to SW).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.03.010.
| [1] | L.K. Tsou, Y. Cheng, Y.C. Cheng. Therapeutic development in targeting proteinprotein interactions with synthetic topological mimetics. Curr. Opin. Pharmacol. 12 (2012) 403–407. DOI:10.1016/j.coph.2012.04.004 |
| [2] | J.M. Rogers, A. Steward, J. Clarke. Folding and binding of an intrinsically disordered protein: fast but not 'diffusion-limited'. J. Am. Chem. Soc. 135 (2013) 1415–1422. DOI:10.1021/ja309527h |
| [3] | J.A. Wells, C.L. McClendon. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450 (2007) 1001–1009. DOI:10.1038/nature06526 |
| [4] | N. Tsomaia. Peptide therapeutics: targeting the undruggable space. Eur. J. Med. Chem. 94 (2015) 459–470. DOI:10.1016/j.ejmech.2015.01.014 |
| [5] | P. Vlieghe, V. Lisowski, J. Martinez, M. Khrestchatisky. Synthetic therapeutic peptides: science and market. Drug Discov. Today 15 (2010) 40–56. DOI:10.1016/j.drudis.2009.10.009 |
| [6] | S. Cory, J.M. Adams. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2 (2002) 647–656. DOI:10.1038/nrc883 |
| [7] | R.J. Youle, A. Strasser. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell. Biol. 9 (2008) 47–59. DOI:10.1038/nrm2308 |
| [8] | K. Wang, X.M. Yin, D.T. Chao, C.L. Milliman, S.J. Korsmeyer. BID: a novel BH3 domain-only death agonist. Genes Dev. 10 (1996) 2859–2869. DOI:10.1101/gad.10.22.2859 |
| [9] | M. Sattler, H. Liang, D. Nettesheim, et al., Structure of Bcl-XL-Bak peptide complex: recognition between regulators of apoptosis. Science 275 (1997) 983–986. DOI:10.1126/science.275.5302.983 |
| [10] | T. Lindsten, A.J. Ross, A. King, et al., The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6 (2000) 1389–1399. DOI:10.1016/S1097-2765(00)00136-2 |
| [11] | E.H.Y.A. Cheng, M.C. Wei, S. Weiler, et al., BCL-2, BCL-XL sequester BH3 domainonly molecules preventing BAX-and BAK-mediated mitochondrial apoptosis. Mol. Cell 8 (2001) 705–711. DOI:10.1016/S1097-2765(01)00320-3 |
| [12] | J.M. Adams. Ways of dying: multiple pathways to apoptosis. Genes Dev. 17 (2003) 2481–2495. DOI:10.1101/gad.1126903 |
| [13] | P.E. Czabotar, G. Lessene, A. Strasser, J.M. Adams. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15 (2014) 49–63. |
| [14] | A. Delbridge, A. Strasser. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 22 (2015) 1071–1080. DOI:10.1038/cdd.2015.50 |
| [15] | C. Billard. BH3 mimetics: status of the field and new developments. Mol. Cancer Ther. 12 (2013) 1691–1700. DOI:10.1158/1535-7163.MCT-13-0058 |
| [16] | D.C.S. Huang, A. Strasser. BH3-only proteins—essential initiators of apoptotic cell death. Cell 103 (2000) 839–842. DOI:10.1016/S0092-8674(00)00187-2 |
| [17] | A.M. Petros, E.T. Olejniczak, S.W. Fesik. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 1644 (2004) 83–94. DOI:10.1016/j.bbamcr.2003.08.012 |
| [18] | M.M. Harris, Z. Coon, N. Alqaeisoom, B. Swords, J.M. Holub. Targeting antiapoptotic Bcl2 proteins with scyllatoxin-based BH3 domain mimetics. Org. Biomol. Chem. 14 (2016) 440–446. DOI:10.1039/C5OB02080H |
| [19] | E.N. Murage, G.Z. Gao, A. Bisello, J.M. Ahn. Development of potent glucagonlike peptide-1 agonists with high enzyme stability via introduction of multiple lactam bridges. J. Med. Chem. 53 (2010) 6412–6420. DOI:10.1021/jm100602m |
| [20] | Y.H. Lau, P. de Andrade, Y.T. Wu, D.R. Spring. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 44 (2015) 91–102. DOI:10.1039/C4CS00246F |
| [21] | L.D. Walensky, A.L. Kung, I. Escher, et al., Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305 (2004) 1466–1470. DOI:10.1126/science.1099191 |
| [22] | M.D. Boersma, H.S. Haase, K.J. Peterson-Kaufman, et al., Evaluation of diverse α/β-backbone patterns for functional α-helix mimicry: analogues of the Bim BH3 domain. J. Am. Chem. Soc. 134 (2012) 315–323. DOI:10.1021/ja207148m |
| [23] | K.J. Peterson-Kaufman, H.S. Haase, M.D. Boersma, et al., Residue-based preorganization of BH3-derived α/β-peptides: modulating affinity, selectivity and proteolytic susceptibility in α-helix mimics. ACS Chem. Biol. 10 (2015) 1667–1675. DOI:10.1021/acschembio.5b00109 |
| [24] | J.W. Checco, E.F. Lee, M. Evangelista, et al., α/β-Peptide foldamers targeting intracellular protein-protein interactions with activity in living cells. J. Am. Chem. Soc. 137 (2015) 11365–11375. DOI:10.1021/jacs.5b05896 |
| [25] | M.D. Boersma, J.D. Sadowsky, Y.A. Tomita, S.H. Gellman. Hydrophile scanning as a complement to alanine scanning for exploring and manipulating protein-protein recognition: application to the Bim BH3 domain. Protein Sci. 17 (2008) 1232–1240. DOI:10.1110/ps.032896.107 |
| [26] | H. Jo, N. Meinhardt, Y. Wu, et al., Development of α-helical calpain probes by mimicking a natural protein-protein interaction. J. Am. Chem. Soc. 134 (2012) 17704–17713. DOI:10.1021/ja307599z |
| [27] | Z.M. Wu, S.Z. Liu, X.Z. Cheng, et al., Recent progress of on-resin cyclization for the synthesis of clycopeptidomimetics. Chin. Chem. Lett. 27 (2016) 1731–1739. DOI:10.1016/j.cclet.2016.04.024 |
| [28] | Y. Li, C.H. Shi, Q.J. Lv, et al., GLP-1 C-terminal structures affect its blood glucose lowering-function. J. Pept. Sci. 14 (2008) 777–785. DOI:10.1002/psc.v14:7 |
| [29] | S. Ottilie, J.L. Diaz, W. Horne, et al., Dimerization properties of human BAD: identification of a BH-3 domain and analysis of its binding to mutant BCL-2 and BCL-XL proteins. J. Biol. Chem. 272 (1997) 30866–30872. DOI:10.1074/jbc.272.49.30866 |
| [30] | J.L. LaBelle, S.G. Katz, G.H. Bird, et al., A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 122 (2012) 2018–2031. DOI:10.1172/JCI46231 |
| [31] | T. Okamoto, K. Zobel, A. Fedorova, et al., Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity. ACS Chem. Biol. 8 (2012) 297–302. |
| [32] | T. Okamoto, D. Segal, K. Zobel, et al., Further insights into the effects of preorganizing the BimBH3 helix. ACS Chem. Biol. 9 (2014) 838–839. DOI:10.1021/cb400638p |
| [33] | C.L. Zhang, Y.Y. Qu, X.Q. Wu, et al., Eco-friendly insecticide discovery via peptidomimetics: design, synthesis, and aphicidal activity of novel insect Kinin analogues. J. Agric. Food Chem. 63 (2015) 4527–4532. DOI:10.1021/acs.jafc.5b01225 |
| [34] | W.Z. Wang, M.L. Li, Z.W. Wei, et al., Bimodal imprint chips for peptide screening: integration of high-throughput sequencing by MS and affinity analyses by surface Plasmon resonance imaging. Anal. Chem. 86 (2014) 3703–3707. DOI:10.1021/ac500465e |