 2017, Vol. 28
2017, Vol. 28 



b Division of Advanced Nanomaterials, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215125, China;
c University of Chinese Academy of Sciences, Beijing 100049, China
With the increase of global energy demand and the attentions to the depletion of fossil energy, the development of large-scale alternatives to fossil fuels becomes more important nowadays [1, 2]. Hydrogen can be used as an energy source for fuel cells with the release of water as the only side product and tremendous potential to decrease greenhouse gas emissions. The advantages of hydrogen as an energy source include: 1) high mass energy density (123 MJ/kg), 2) facile conversion to electrical energy as well as mechanical energy, and 3) generation of only water upon conversion to energy. Approximately 50 million metric tons of hydrogen produced from fossil fuels annually are used mainly for oil refining and ammonia synthesis. However, current thermochemical production options are prohibitively expensive [3]. To unlock the tremendous potential of the hydrogen economy, it is highly important to develop an efficient and practical method for the hydrogenation from renewable and easy-to-handle resources.
In recent years, one-carbon molecules constantly arise as good carriers of renewable hydrogen [4-10]. CH3OH is a high hydrogen storage medium (12.5 wt% H2) and a convenient chemical feedstock to produce a myriad of chemicals and products [7, 11-14]. It is already one of the most important building blocks in the chemical industry with an annual production in excess of 70 million tons. Furthermore, it can be directly used as a drop-in liquid fuel for internal combustion engines and direct methanol fuel cells (DMFC). Besides, compared with the difficulties of selective activation of methane and the inferior energy balance of formic acid, methanol is the most promising one-carbon molecule in the world currently [8, 15-20].
There are mainly three methods for hydrogen production from methanol, that is, methanol steam reforming for hydrogen production (MSR), partially catalytic oxidation of methanol reforming (POR), and auto-thermal reforming of methanol (ATR). Among them, the MSR is the most economic method, by which the molar ratio of H2 and CO2 in gaseous product is about 3:1 theoretically. The so-called methanol-reforming process allows for the production of H2 from a mixture of methanol and water. However, limited by the catalytic activity and selectivity of the reported catalysts, the MSR always perform at high temperatures ( > 200 ℃). In addition, the co-generation of carbon monoxide limits its further applications in fuel cells which only bear a low CO concentrations [7, 21]. Moreover, the subsequent purification of the reforming gas would increase the costs of hydrogen generation. To meet the requirements of applications and reducing cost, catalysts with high activity and selectivity are needed.
Herein, we described an aqueous phase methanol reforming process under low temperatures ( < 100 ℃) and mild conditions. With the existence of homogeneous rhodium catalyst, no carbon monoxide could be detected in the reforming gas. Hydrogen generation was observed with excellent turnover frequencies (TOF) of 83.2 h-1 by [Cp*Ru(NH3)(H2O)2]3+ at 70 ℃ without any addition of alkaline or organic materials.
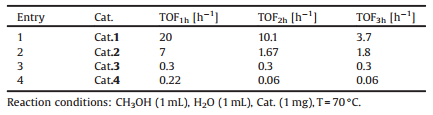
2. Results and discussionSeveral organo-rhodium complex catalysts were tested for the methanol reforming reaction. All the experiments were carried out at 70 ℃. The activities of various catalysts for methanol reforming were listed in Table 1. The chemical equation has been confirmed as follows:
|
|
Table 1 Catalytic activity of methanol reforming for hydrogen production by Rh catalysts bearing different ligands. |

|
(1) |
As shown in Fig. 1, different from other reported catalysts which bear the pincer-type ligands [22-25], the central rhodium of the four catalysts all bears one Cp* ligand, and ligands on the remaining empty coordination numbers are different from each other. Three null coordination of cat.1 are filled with one NH3 and two H2O molecules, respectively. 4-(1H-Imidazol-1-yl) benzoic acid relies on the nitrogen of the imidazolyl to connect with Rh-Cp*, and the rest of the coordination is filled with one water molecule. N, N-Chelate bond were generated by the Rh-Cp* and ligands of cat.3 and cat.4. 2, 2'-Bipyrimidine is connected to rhodium by two nitrogen on the same side, and water molecule acts as another ligand. The 2, 2'-biimidazole and 2, 2'-bipyrimidine are all chelating reagent, which could connect to Rh-Cp* strongly.

|
Download:
|
| Fig. 1. Corresponding structures of the four catalysts. | |
{kind=link}
As we all know, it is very difficult to break N, N-chelate bond of the catalysts, which as a result, greatly hampered the hydrogen evolution [26, 27]. Table 1 shows that the TOF values of cat.3 and cat.4 were below 0.5, which was determined by their similar structures. The chelating ligand could increase the steric hindrance during the catalytic reaction [28, 29]. The catalytic activity of cat.2 increased to 7-8 times compared to the former two. Remarkably, the catalytic activity of cat.1 with NH3 as a ligand quickly upgraded to about 20 times, and TOF value reached 20 h-1. Apparently, the difference in activities lies in the difference of coordination environment. Catalysts with multiple active sites can greatly enhance the catalytic activity, and synchronously, reactants are more likely to be exposed to the active transition metal. In general, the catalytic activity decreased with the increase of time for the methanol dehydrogenation.
Effects of reaction temperatures, molar ratio of water to methanol and reaction time on the performance of the reaction should be investigated. The concentration of carbon monoxide in the reforming gas has a strongly negative effect on the efficiency of proton exchange membrane fuel cell (PEMFC). Optimization of the reaction conditions is beneficial to improve the conversion of the reactant and the selectivity of the product. After demonstrating the reactivity of the four catalysts for methanol reforming, cat.1 was chosen to assess the influence of the reaction conditions (pH, temperature and water content) on the performance of the catalyst.
As can be seen from Fig. 2a, when the ratio of methanol to water was between 1:4.5-3:1, the reaction activity gradually increased with the increase of the ratio, namely, the methanol conversion rate was gradually increased. There is a sharp decrease upon increasing the ratio to 5:1. Overall, the catalytic activity decreased over time. Fig. 2b shows the effect of the initial methanol to water ratio on the initial dehydrogenation reactivity (TOF1h). In brief, 3:1 was the optimum ratio of methanol to water for methanol dehydrogenation. The reaction mechanism of the methanol dehydrogenation was proposed and discussed as follows:

|
Download:
|
| Fig. 2. (a) Effect of the initial water methanol ratio on the hydrogen evolution with time increase. (b) Effect of the initial water methanol ratio on the TOF1h. T = 70 ℃. | |
{kind=link}

|
(2) |

|
(3) |
For the first step, the process of C-H bonds of methanol breaking into hydrogen and formaldehyde was an up-hill reaction, which made the methanol dehydrogenation very difficult and determined the whole reaction rate. The following step (3) was the thermodynamically down-hill reaction, which could be carried out quickly in the presence of catalysts. It was also the renowned "aldehyde-water shift (AWS)" reaction, in which water played the role of the effective oxidant [30, 31]. During this process, the aldehyde group was firstly oxidized into carboxyl group, meanwhile, releasing hydrogen as the valuable by-product. Subsequently, formic acid was decomposed into hydrogen and carbon dioxide rapidly [32, 33]. Obviously, as the reactant and oxidant, water content had a great influence on the reaction rate.
The addition of some basic or organic substances into the system were typically used to accelerate the reaction rate, especially for the first step [7, 34, 35]. Considering the high requirements on the container and delivery process caused by alkaline and organic materials, aqueous solution without extra alkaline or organic substances is desired for industrial applications nowadays [36, 37]. The reaction systemwe described here can meet the above requirements well.
We used GC to measure the composition of the evolved gas from the methanol reforming reaction. No CO was detected (below detection limit) in the generated gas (Fig. 3c). Besides hydrogen, the only detectable gas was CO2 (Fig. 3a and b). Hydrogen generation without CO is especially useful to the high performance of PEMFC.

|
Download:
|
| Fig. 3. GC spectra of the gaseous product from methanol reforming reaction. (a) H2; (b) CO2; (c) CO. Orange curves represent the standard gas samples. Reaction conditions: methanol (1.71 mL), deionized H2O (0.29 mL), cat.1 (3.4 μmol), T = 70 ℃. The evolved gas for gas chromatogram measurement was collected after reacting 1 h. | |
{kind=link}
Fig. 4a shows thathigher temperature couldincrease the reaction rate. The TOF value reached up to 20 without any additional material required at 343 K. Moreover, the apparent activation energy (Ea) could be achieved by fitting the linear correlation between ln(TOF) and 1/T accurately. The Ea for methanol reforming was 96.0 kJ/mol. As exhibited in Fig. 4b, the effect of buffer pH on the methanol dehydrogenation was observed in the pH range from 5 to 8, which were adjusted by phosphate buffer solution. At such relatively low temperatures, the hydrogen evolution rate increased gradually with elevating the buffer pH from 5 to 6 and presented a maximum TOF of 83.2 h-1 at pH 6. Further increasing the pH to 8 dramatically decreased the reaction rate.

|
Download:
|
| Fig. 4. (a) Effect of the temperature on the catalytic hydrogen generation; (b) Effect of the initial buffer pH on the hydrogen evolution. Reaction conditions: (a) methanol (1.71 mL), deionized H2O (0.29 mL), cat.1 (3.4 μmol). (b) Methanol (1.71 mL), deionized H2O (0.19 mL), buffer solution (20 mmol/L, 0.1 mL), cat.1 (3.4 μmol), T = 70 ℃. | |
{kind=link}
3. Conclusion
In summary, we developed an unprecedented mild reaction system for methanol reforming by [Cp*Rh(NH3)(H2O)2]3+ in aqueous phase, and no additional alkaline or organic substances are required. Comparison of several different catalysts revealed that the coordinative H2O ligand is labile, and the left vacant site allows the metal center for catalysis, which finally accelerates the reaction rate. Hydrogen with low CO content was obtained under 70 ℃ and constant pressure with TOF of 83.2 h-1. The optimum mole ratio of methanol to water was 3:1, the pH of buffer had a great effect on the reforming process, and faintly acidic conditions were conducive to the reaction. Similarly, alkaline conditions counted against the reaction. Reacting below 70 ℃ opens the possibility to perform aqueous methanol reforming with the waste heat from proton-exchange membrane fuel cells. Furthermore, no CO was detected in the product gas, which means that the gaseous product can be directly used for fuel cells. In addition, slightly acidic conditions can avoid the corrosion to the container caused by the strong acid or alkaline solution, which extends its application range and makes it capable to meet the requirements of industrial application. These findings are very beneficial for the future industrial application of hydrogen production.
4. Experimental 4.1. Materials and instruments2, 2'-Bipyrimidine were obtained from J & K Chemical Co., (Shanghai, China). 2, 2'-Biimidazole were purchased from Alfa Aesar. Cp* was provided by Meryer (Shanghai) Chemical Technology Co. 4-(1H-Imidazol-1-yl) benzoic acid was obtained from Energy (Shanghai) Chemical Co. Other chemicals were obtained from China National Pharmaceutical Group Co. (Shanghai, China). All chemicals are analytical reagents. All commercial materials were used as received unless specified.
The gaseous product generated from low temperature methanol reforming was analyzed by GC-G5 (Beijing Persee General Instrument Co., Ltd.), the gas chromatography assembled with TCD, FID, TDX-1 column and methane conversion reactor. N2 was used as carrier gas. The detection limit of CO was below 10 ppm through a methane reformer. TCD of the GC was used to detect H2. The 1H NMR spectrums were characterized by NMR (Varian Plus 400 MHz). The UV-vis spectrum was collected by Shimadzu UV 1800.
4.2. Synthesis of the four catalystsSynthesis of [Cp*Rh(NH3)(H2O)2]3+ (cat.1): 200 mg [RhCp*Cl2]2 was stirred in 15 mL water at room temperature, forming a pale yellow suspension. Then 200 mL ammonia was dropped into the suspension slowly. Keep stirring at room temperature for 8 h, the solid will dissolve and turn into dark red solution. Then reddish brown product will be gained by evaporating solution under reduced pressure. Synthesis process refers to literature partially [38]. The corresponding 1H NMR spectra is shown in Fig. S3 in Supporting information. 1H NMR (400 MHz, D2O): δ 1.60 (s, 15H), 1.19 (t, 3H).
Synthesis of cat.2: An aqueous solution of [RhⅢ(Cp*)(H2O)3](SO4) and equiv. of 4-(1H-pyrazol-1-yl)benzoic acid was stirred under reflux for 12 h, and then the solution was filtered with a membrane filter. The filtrate was evaporated under reduced pressure to yield a yellow powder of cat.2 [39]. 1H NMR spectra is shown in Fig. S4 in Supporting information. 1H NMR (400 MHz, DMSO-d6): δ 0.35 (s, 15H), 5.56 (s, 1H), 6.44 (s, 1H), 6.66 (s, 1H), 6.79 (s, 1H), 6.84 (s, 1H), 7.13 (s, 1H), 7.48 (s, 1H).
Synthesis of cat.3: Reaction of the rhodium dimers [{(Cp*)RhCl2}2] with 2 equiv. of biimidazole yielded the cationic complexes, which were isolated as chloride salts. The yellow compounds are air-stable, soluble in water and in most organic solvents [40]. The corresponding 1H NMR spectra is shown in Fig. S5 in Supporting information. 1H NMR (400 MHz, D2O): δ 1.76 (s, 15H), 7.29 (s, 2H), 7.47 (s, 2H), 7.56 (s, 2H).
Synthesis of cat.4: The pentamethylcyclopentadienyl complexes [Cp*RhCl2]2 react with 2 equiv. of 2, 2'-bipyrimidine (bpym) in methanol to form the cationic pentamethylcyclopentadienyl complexes [(η5-C5Me5) RhCl(bpym)]+ [23]. 1H NMR spectra is shown in Fig. S6 in Supporting information. 1H NMR (400 MHz, D2O): δ 1.64 (s, 15H), 7.96 (t, 2H), 9.20 (m, 4H).
4.3. General experimental proceduresA small stirring magnet was put into the clean round-bottom flask beforehand, and then the pre-configured reaction solution was added into the flask and the flask was sealed with a rubber stopper. Next, the round-bottom flask was heated in a water bath at a pre-set temperature (70 ℃) under ambient atmosphere. After a period of time, a certain amount of catalyst solution was injected into the reactor through a micro syringe. A semi liquid filled Ushaped tube was connected to the reactor. The volume content of the evolved gas could be calculated through changes of liquid level in U-shaped tube. A camera was used to monitor the changes of liquid level. The schematic diagram of the process is shown in Fig. S1 in Supporting information.
4.4. Calculation of TOF and the volume of H2TOF was calculated by Eq. (4) [41].

|
(4) |
The calculation of Vm, H2, 20 ℃ was carried out using van der Waals Eq. (5); Vtotal is the total amount of gas produced; t is the reaction time required to produce these gases; ncatalyst is the molar quantities of catalyst.

|
(5) |
where R: 8.3145 m3 Pa/mol/K; T: 293.15 K; P: 101325 Pa; b: 26.7 × 10-6 m3/mol; a: 2.49 × 10-10 Pa m3/mol2.
AcknowledgmentsThe authors are grateful for financial support granted by Ministry of Science and Technology of the People's Republic of China (Nos. 2016YFA0200700 and 2016YFE0105700), the National Natural Science Foundation of China (Nos. 21373264 and 21573275), the Natural Science Foundation of Jiangsu Province (No. BK20150362), Suzhou Institute of Nano-tech and Nanobionics (No. Y3AAA11004) and Thousand Youth Talents Plan (No. Y3BQA11001).
| [1] | B.C. Steele, A. Heinzel. Materials for fuel-cell technologies. Nature 414 (2001) 345–352. DOI:10.1038/35104620 |
| [2] | S. Sahler, M.H. Prechtl. Advancement in molecular hydrogen storage systems. Chem. Cat. Chem. 3 (2011) 1257–1259. |
| [3] | L. Schlapbach, A. Zuttel. Hydrogen-storage materials for mobile applications. Nature 414 (2001) 353–358. DOI:10.1038/35104634 |
| [4] | A. Boddien, B. Loges, F. Gaertner, et al., Iron-catalyzed hydrogen production from formic acid. J. Am. Chem. Soc. 132 (2010) 8924–8934. DOI:10.1021/ja100925n |
| [5] | B. Loges, A. Boddien, H. Junge, et al., Controlled generation of hydrogen from formic acid amine adducts at room temperature and application in H2/O2 fuel cells. Angew. Chem. Int. Ed. 47 (2008) 3962–3965. DOI:10.1002/anie.v47:21 |
| [6] | R. Tanaka, M. Yamashita, K. Nozaki. Catalytic hydrogenation of carbon dioxide using Ir(Ⅲ)-pincer complexes. J. Am. Chem. Soc. 131 (2009) 14168. DOI:10.1021/ja903574e |
| [7] | M. Nielsen, E. Alberico, W. Baumann, et al., Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide. Nature 495 (2013) 85–89. DOI:10.1038/nature11891 |
| [8] | C. Fellay, P.J. Dyson, G. Laurenczy. A viable hydrogen-storage system based on selective formic acid decomposition with a ruthenium catalyst. Angew. Chem. Int. Ed. 47 (2008) 3966–3968. DOI:10.1002/anie.v47:21 |
| [9] | P. Hu, Y. Diskin-Posner, Y. Ben-David, et al., Reusable homogeneous catalytic system for hydrogen production from methanol and water. ACS Catal. 4 (2014) 2649–2652. DOI:10.1021/cs500937f |
| [10] | R.E. Rodriguez-Lugo, M. Trincado, M. Vogt, et al., A homogeneous transition metal complex for clean hydrogen production from methanol-water mixtures. Nat. Chem. 5 (2013) 342–347. DOI:10.1038/nchem.1595 |
| [11] | S. Wesselbaum, T. vom Stein, J. Klankermayer, et al., Hydrogenation of carbon dioxide to methanol by using a homogeneous ruthenium-phosphine catalyst. Angew. Chem. Int. Ed. 51 (2012) 7499–7502. DOI:10.1002/anie.201202320 |
| [12] | G.M. Meaburn, F.W. Mellows, A. Reiffste. Production of hydrogen in radiolysis of methanol vapour. Nature 204 (1964) 1301. DOI:10.1038/2041301a0 |
| [13] | G.A. Olah. Beyond oil and gas: the methanol economy. Angew. Chem. Int. Ed. 44 (2005) 2636–2639. DOI:10.1002/(ISSN)1521-3773 |
| [14] | G.A. Olah. Jenseits von Öl und gas: die methanolwirtschaft. Angew. Chem. 117 (2005) 2692–2696. DOI:10.1002/ange.200462121 |
| [15] | G.A. Olah. Towards oil independence through renewable methanol chemistry. Angew. Chem. Int. Ed. 52 (2013) 104–107. DOI:10.1002/anie.201204995 |
| [16] | G.A. Olah. Der Weg in die Unabhängigkeit vom Öl mithilfe einer chemie auf der basis von erneuerbarem methanol. Angew. Chem. 125 (2013) 112–116. DOI:10.1002/ange.201204995 |
| [17] | M.H.G. Prechtl, S. Sahler. Hydrogen storage using ionic liquid media. Curr. Org. Chem. 17 (2013) 220–228. DOI:10.2174/1385272811317030004 |
| [18] | A. Boddien, C. Federsel, P. Sponholz, et al., Towards the development of a hydrogen battery. Energy Environ. Sci. 5 (2012) 8907–8911. DOI:10.1039/c2ee22043a |
| [19] | M. Grasemann, G. Laurenczy. Formic acid as a hydrogen source—recent developments and future trends. Energy Environ. Sci. 5 (2012) 8171–8181. DOI:10.1039/c2ee21928j |
| [20] | A. Boddien, D. Mellmann, F. Gaertner, et al., Efficient dehydrogenation of formic acid using an iron catalyst. Science 333 (2011) 1733–1736. DOI:10.1126/science.1206613 |
| [21] | D.R. Palo, R.A. Dagle, J.D. Holladay. Methanol steam reforming for hydrogen production. Chem. Rev. 107 (2007) 3992–4021. DOI:10.1021/cr050198b |
| [22] | R.Y. Abrokwah, V.G. Deshmane, D. Kuila. Comparative performance of MMCM-41(M: Cu, Co, Ni, Pd, Zn and Sn) catalysts for steam reforming of methanol. Inorg. Chem. 425 (2016) 10–20. |
| [23] | J. Canivet, L. Karmazin-Brelot, G. Suss-Fink. Cationic arene ruthenium complexes containing chelating 1, 10-phenanthroline ligands. J. Organomet. Chem. 690 (2005) 3202–3211. DOI:10.1016/j.jorganchem.2005.02.050 |
| [24] | W. Zhou, Y. Ke, Q. Wang, et al., Development of cylindrical laminated methanol steam reforming microreactor with cascading metal foams as catalyst support. Fuel 191 (2017) 46–53. DOI:10.1016/j.fuel.2016.11.058 |
| [25] | D. Liu, Y. Men, J. Wang, et al., Highly active and durable Pt/In2O3/Al2O3 catalysts in methanol steam reforming. Int. J. Hydrogen Energy 41 (2016) 21990–21999. DOI:10.1016/j.ijhydene.2016.08.184 |
| [26] | N.A. Gothard, M.W. Mara, J. Huang, et al., Strong steric hindrance effect on excited state structural dynamics of Cu(Ⅰ) diimine complexes. J. Phys. Chem. A 116 (2012) 1984–1992. |
| [27] | H. Gao, L. Pei, Y. Li, et al., Vinyl polymerization of norbornene with nickel catalysts bearing N, N six-membered chelate ring: important influence of ligand structure on activity. J. Mol. Catal. A: Chem. 280 (2008) 81–86. DOI:10.1016/j.molcata.2007.10.033 |
| [28] | C. Zuccaccia, A. Macchioni, I. Orabona, et al., Interionic solution structure of [PtMe(η2-olefin)(N, N-diimine)]BF4 complexes by 19F{1H}-HOESY NMR spectroscopy: effect of the substituents on the accessibility of the counterion to the metal. Organometallics 18 (1999) 4367–4372. DOI:10.1021/om990401r |
| [29] | R. Scopelliti, G. Bruno, C. Donato, et al., Incorporation of non-planar chelating ligands in the coordination sphere of ruthenium(Ⅱ) complexes-unusual S-thioether N-pyridyl chelation mode of di-2-pyridyl sulfide (dps) to Ru(N, N-dps)2 core: NMR studies of sterically induced internal dynamics. Inorg. Chim. Acta 313 (2001) 43–55. DOI:10.1016/S0020-1693(00)00343-1 |
| [30] | T.P. Brewster, W.C. Ou, J.C. Tran, et al., Iridium, Rhodium, and ruthenium catalysts for the aldehyde-water shift reaction. ACS Catal 4 (2014) 3034–3038. DOI:10.1021/cs500843a |
| [31] | L.E. Heim, N.E. Schloerer, J.H. Choi, et al., Selective and mild hydrogen production using water and formaldehyde. Nat. Commun. 5 (2014) . |
| [32] | J.H. Sinfelt, D.J. Yates. Catalytic hydrogenolysis of ethane over the noble metals of Group Ⅷ. J. Catal. 8 (1967) 82–90. DOI:10.1016/0021-9517(67)90284-9 |
| [33] | N. Taccardi, D. Assenbaum, M.E.M. Berger, et al., Catalytic production of hydrogen from glucose and other carbohydrates under exceptionally mild reaction conditions. Green Chem. 12 (2010) 1150–1156. DOI:10.1039/c002910f |
| [34] | M. Yadav, Q. Xu. Liquid-phase chemical hydrogen storage materials. Energy Environ. Sci. 5 (2012) 9698–9725. DOI:10.1039/c2ee22937d |
| [35] | T. He, P. Pachfule, H. Wu, et al., Hydrogen carriers. Nat. Rev. Mater. 1 (2016) 16059. DOI:10.1038/natrevmats.2016.59 |
| [36] | H. Junge, A. Boddien, F. Capitta, et al., Improved hydrogen generation from formic acid. Tetrahedron Lett. 50 (2009) 1603–1606. DOI:10.1016/j.tetlet.2009.01.101 |
| [37] | G.S. Coumbarides, J. Eames, M. Motevalli, et al., (-)-Dimenthyl malonate. Acta Crystallogr. C 58 (2002) 1084–1085. |
| [38] | P.A. Lay, R.H. Magnuson, H. Taube. Sythesis of N-heterocyclic complexes of osmium, ruthenium, cobalt, and rhodium pentaammines. Inorg. Chem. 27 (1988) 2848–2853. DOI:10.1021/ic00289a022 |
| [39] | M.T.Y. Raymond Ziessel. Fadila Balegroune, Daniel Grandjean, Synthesis and molecular structure of new families oriridium(Ⅲ)-Cp* and rhodium(Ⅲ)-Cp* complexes derived from 1, 2-dicyanoethene-1, 2-dithiolate, 2, 2'-biimidazole or 2. 2'-bithiazole. Single crystal structures of [η5-C5Me5 Ir(biimH2)Cl]Cl and [(η5-C5Me5)Rh(dcdt)]. J. Organomet. Chem. 441 (1992) 143–154. DOI:10.1016/0022-328X(92)80198-7 |
| [40] | P. Govindaswamy, J. Canivet, B. Therrien, et al., Mono and dinuclear rhodium, iridium and ruthenium complexes containing chelating 2, 2'-bipyrimidine ligands: synthesis, molecular structure, electrochemistry and catalytic properties. J. Organomet. Chem. 692 (2007) 3664–3675. DOI:10.1016/j.jorganchem.2007.04.048 |
| [41] | Z. Wang, S.-M. Lu, J. Li, et al., Unprecedentedly high formic acid dehydrogenation activity on an iridium complex with an N, N'-diimine ligand in water. Chem. Eur. J. 21 (2015) 12592–12595. DOI:10.1002/chem.201502086 |