 2017, Vol. 28
2017, Vol. 28 



b School of Materials and Chemical Engineering, Anhui Jianzhu University, Hefei 230022, China
Gold nanoparticles (AuNPs) have long been a topic of intense research due to their size-related electronic, magnetic and optical properties (quantum size effect), and these properties made them have potential applications in catalysis, nanoelectronic devices, and highly sensitive chemical or biological sensors [1-6]. The synthesis of gold nanoparticles, usually involves the incorporation of matrices such as surfactants, copolymers, or dendrimers, which act as stabilizers in order to avoid agglomeration [7-9]. As a fascinating branch, AuNPs formation in the presence of block copolymer micellar templates is additionally attractive due to colloidally stable nanoparticles of controlled size and chemical functionality in the final hybrid nanocolloids.
In principle, the interparticle distances within the AuNPs assemblies greatly relevant to their electronic and magnetic properties can be modulated by tuning the incorporation positions at the polymer micellar scaffolds, e.g. the inner core, the outer shell, or the outer periphery. For example, Alexandridis and coworkers showed that poly(ethylene oxide)-b-poly(propylene oxide) (PEOb-PPO) block copolymer could act as the reducing, stabilizing, and morphogenic agent at the one pot synthesis of Au nanoparticles where the micellar cores of amphiphilic block copolymers are regarded as nanoreactors for the nucleation and growth of AuNPs. Furthermore, they reported the size and shape controlled synthesis of the colloidal AuNPs [10, 11]. The strategy concerning the formation of AuNPs into the micellar cores has the deficiencies that some properties of gold nanoparticles (i.e. catalytic) may be inhibited or compromised because they are embedded in the dense micellar core. To overcome this issue, Shi et al. fix the Au nanoparticles on the coronas of the micelles consisting of a poly (ethylene oxide) core and a swollen poly(4-vinylpyridine) corona sensitive to pH, allowing for interaction with biological molecules [12]. In our previous studies, gold nanoparticles were covalently conjugated onto the periphery of thiol functionalized thermosensitive poly(N-isopropylacrylamide) (PNIPAM) unimolecular micelles in order to fabricate satellite-like nanostructures. These micellar templates were able to shrink and swell reversibly in response to thermal treatment. Consequently, the spatial distances between gold nanoparticles attached at the micelles surface, could be altered [13].
Despite tremendous progresses, however, it remains challengeable to delicately modulate the spatial locations of gold nanoparticles in the assemblies (e.g. core/shell interface). The core/shell boundary has been observed to give rise to interesting physical and chemical properties exhibiting important technological applications [14-17]. Due to the dense alignments and chemical potentials of the interfacial regions, we anticipated, therefore, that the properties of gold nanoparticle anchored at the core/shell interface could be remarkably different to the pure metal nanoparticles.
Herein, we combined atom transfer radical polymerization (ATRP) and reversible addition-fragmentation chain transfer (RAFT) polymerization to synthesis cleavable ABA triblock copolymers from individual homopolymer blocks. Firstly, we synthesized homopolymers with one or two terminals of pyridyldisulfide (PyDS) groups, PyDS-PMMA and PyDS-PEO-PyDS, and with one or two terminals of thiol groups, PDEA-SH and HSPMEO3MA-SH, where PMMA, PEO, PDEA, and PMEO3MA represent poly(methyl methacrylate), poly(ethylene oxide), poly(2-(diethylamino)ethyl methacrylate), and poly(tri(ethylene glycol) monomethyl ether methacrylate, respectively. Then, by substitution reaction between PyDS-PMMA and HS-PMEO3MA-SH, or between PyDS-PEO-PyDS and PDEA-SH, we prepared two ABA triblock copolymers with disulfide as the junction point. The synthesized triblock copolymers could self-assemble into spherical micelles in aqueous solutions. The micelles had insoluble cores, hydrophilic outer shells, and a large amount of disulfide groups in the interface between the cores and shells. These disulfide groups were then used to precisely load gold nanoparticles with ~2 nm in diameter at the core/shell interface (Scheme 1), and the detail structures of the gold nanoparticles incorporated hybrid micelles were investigated by dynamic laser light scattering (DLS) and transmission electron microscopy (TEM) measurements.

|
Download:
|
| Scheme1. Schematic illustration of polymeric micelles self-assembly from PMMA36-ss-PMEO3MA90-ss-PMMA36 or PDEA36-ss-PEO104-ss-PDEA36 triblock copolymers and the fabrication of gold nanoparticle incorporated hybrid micelles. | |
{kind=link}
2. Experimental 2.1. Materials and characterization
2-(Diethylamino) ethyl methacrylate (DEA), methyl methacrylate (MMA), tri(ethylene glycol) monomethyl ether methacrylate (MEO3MA) were purchased from Aldrich. They were passed through basic alumina columns, then vacuum-distilled from CaH2, and stored at -20 ℃ prior to use. Poly(ethylene oxide) (HO-PEO104-OH, Mn = 5000, Mw/Mn = 1.1, mean degree of polymerization, DP, is 104) was purchased from Fluka and used without further purification. 2, 2'-Dithiodipyridine, 2-mercaptoethanol, tetrakis(hydroxymethyl)phosphonium chloride (THPC, 80% solution in water), and N, N, N', N', N"-pentamethyldiethylenetriamine (PMDETA) were purchased from Sigma-Aldrich and used as received. Succinic anhydride and 2, 2'-azoisobutyronitrile (AIBN) were purified by recrystallization from toluene and 95% ethanol prior to use, respectively. Water was deionized with a Milli-Q SP reagent water system (Millipore) to a specific resistivity of 18.4 MV cm. N, N'-Dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP) and all other reagents were purchased from Sinopharm Chemical Reagent Co., Ltd. and used as received unless otherwise noted. 2-(Pyridin-2-yldisulfanyl)ethan-1-ol (PyDS-OH) [18], 2-(pyridin-2-yldisulfanyl)ethyl 2-bromo-2-methylpropanoate (PyDS-Br) [18], ethane-1, 2-diyl bis(4-cyano-4-((phenylcarbonothioyl)thio)pentanoate) (RAFT agent 1) [19], 2-cyanoprop-2-yl dithiobenzoate (CPDB) [20], and THPC-stabilized gold nanoparticles (~2 nm in diameter) [21] were synthesized according to literature procedures.
Molecular weights and molecular weight distributions were determined by gel permeation chromatography (GPC) equipped with Waters 1515 pump and Waters 2414 differential refractive index detector (set at 45 ℃). It used a series of two linear Styragel columns (HR2 and HR4) at an oven temperature of 45 ℃. The eluent was THF at a flow rate of 1.0 mL/min. A series of low polydispersity polystyrene standards were employed for calibration. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AV300 NMR spectrometer (resonance frequency of 300 MHz for 1H and 75 MHz for 13C) operated in the Fourier transform mode. CDCl3 was used as the solvent. The degrees of polymerization, DP, of synthesized polymers were determined by 1H NMR analysis. Dynamic laser light scattering (DLS) measurements were conducted on a commercial spectrometer (ALV/DLS/ SLS-5022F) equipped with a multi-tau digital time correlator (ALV5000) and a cylindrical 22 mW UNIPHASE He-Ne laser (λ0 = 632 nm) as the light source. Scattered light was collected at a fixed angle of 90° for a duration of 15 min. Transmission electron microscopy (TEM) measurements were conducted on a JEOL 2010 electron microscope. The samples for TEM observations were prepared by dropping 10mL of aqueous dispersions of assemblies onto copper grids successively coated with thin films of Formvar and carbon.
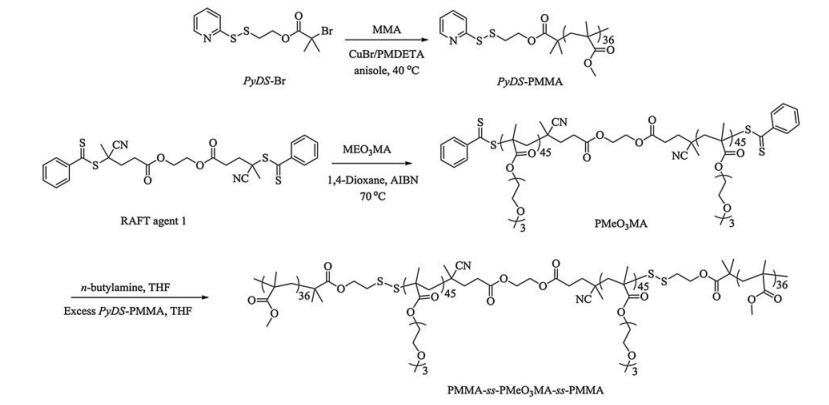
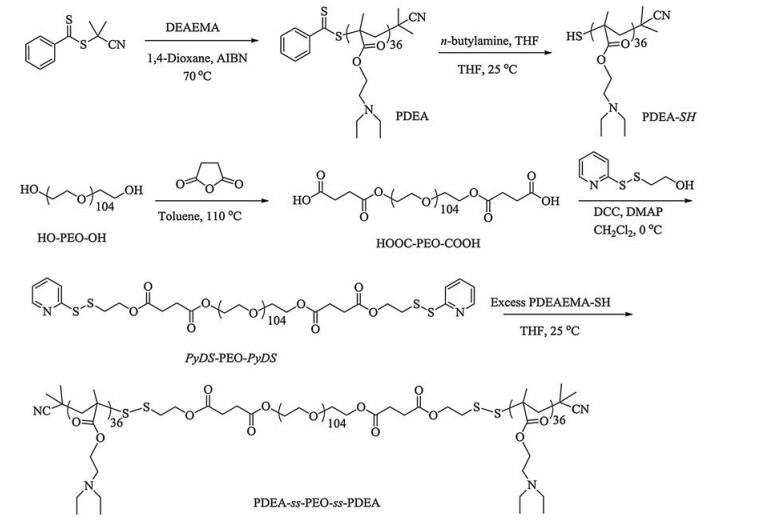
2.2. Sample synthesisThe general approaches to the preparation of cleavable ABA triblock copolymers, PMMA36-ss-PMEO3MA90-ss-PMMA36 and PDEA36-ss-PEO104-ss-PDEA36, are shown in Schemes 2 and 3, respectively.

|
Download:
|
| Scheme2. Synthetic routes employed for the preparation of cleavable ABA triblock copolymer, PMMA36-ss-PMEO3MA90-ss-PMMA36. | |
{kind=link}

|
Download:
|
| Scheme3. Synthetic routes employed for the preparation of cleavable ABA triblock copolymer, PDEA36-ss-PEO104-ss-PDEA36. | |
{kind=link}
2.2.1. Synthesis of PMMA36-ss-PMEO3MA90-ss-PMMA36 triblock copolymer (Scheme 2)
Typical procedures employed for ATRP synthesis of PyDS-PMMA36 were as follows. Briefly, ATRP initiator PyDS-Br (0.068 g, 0.2 mmol), MMA (1.38 g, 13.78 mmol), PMDETA (78 mg, 0.45 mmol) and anisole (3 mL) were charged into a reaction tube equipped with a magnetic stirring bar. The tube was carefully degassed by three freeze-pump-thaw cycles. Before warming to room temperature, CuBr (36 mg, 0.25 mmol) was added into the tube and then the tube was sealed under vacuum. After thermostating at 40 ℃ in an oil bath and stirring for 8 h, the reaction tube was quenched into liquid nitrogen, opened, and diluted with THF, and then the reaction mixture was stirred under air for 4 h. The solution was then subjected to passing through a column of neutral alumina to remove the copper catalysts. After removing all the solvent under reduced pressure, the obtained polymer was dissolved in THF and precipitated into methanol twice. The resulting product was dried overnight under vacuum at room temperature to give PyDS-PMMA as a white solid (1.2 g, yield: 85%). The molecular weight and molecular weight distribution of PyDS-PMMA was determined by GPC using THF as the eluent, revealing an Mn of 6.5 kDa and Mw/Mn of 1.07 (Fig. 1a). The actual degree of polymerization (DP) of the PyDS-PMMA homopolymer was determined to be 36 by 1H NMR analysis recorded in CDCl3 and the obtained homopolymer was denoted as PyDS-PMMA36 (Fig. S1a).

|
Download:
|
| Fig. 1. THF GPC traces obtained for (a) PMEO3MA90, PyDS-PMMA36 homopolymers and disulfide-linked ABA triblock copolymer of PMMA36-ss-PMEO3MA90-ss-PMMA36, (b) PyDS-PEO104-PyDS, PDEA36 homopolymers, and disulfide-linked ABA triblock copolymer of PDEA36-ss-PEO104-ss-PDEA36, (c) PMMA36-ss-PMEO3MA90-ss-PMMA36 copolymers before and after treated with an excess of DTT, and (d) PDEA36-ss-PEO104-ss-PDEA36 copolymers before and after treated with an excess of DTT. | |
{kind=link}
Next, PMEO3MA90 was synthesized through RAFT polymerization as follows. MEO3MA (2.35 g, 10.1 mmol), RAFT agent 1 (54 mg, 0.093 mmol), and AIBN (3 mg, 0.018 mmol) were dissolved in 1, 4-dioxane (2 mL) with a magnetic stirring bar. The tube was carefully degassed by three freeze-pump-thaw cycles and then sealed under vacuum. After thermostating at 70 ℃ in an oil bath and stirring for 16 h, the reaction tube was quenched into liquid nitrogen and opened, and the reaction mixture was then precipitated into an excess of diethyl ether. The above dissolution-precipitation cycle was repeated three times. PMEO3MA was obtained as a pale red liquid (2 g, yield: 85%). The molecular weight and molecular weight distribution of PMEO3MA homopolymer were determined by GPC using THF as the eluent, revealing an Mn of 18.5 kDa and Mw/Mn of 1.13 (Fig. 1a). The actual degree of polymerization (DP) of the PMEO3MA homopolymer was determined to be 90 by 1H NMR analysis recorded in CDCl3 and the obtained homopolymer was denoted as PMEO3MA90 (Fig. S1b).
Finally, PMEO3MA90 (500 mg, 0.024 mmol) and n-butylamine (11 mg, 0.14 mmol) were dissolved in THF. The reaction proceeded under nitrogen for 2 h until the solution color changed from light red to yellow. After the solvent and excess n-butylamine were removed under nitrogen, pre-degassed PyDS-PMMA36 (283 mg, 0.072 mmol) in THF and 20 μL of glacial acetic acid was added, and the reaction mixture was stirred at room temperature under nitrogen for another 24 h. The resulting polymer was precipitated into an excess of diethyl ether three times to give target triblock copolymer PMMA36-ss-PMEO3MA90-ss-PMMA36 (379 mg, yield: 55%). The molecular weight and molecular weight distribution of PMMA36-ss-PMEO3MA90-ss-PMMA36 were determined by GPC using THF as the eluent, revealing an Mn of 33.1 kDa and Mw/Mn of 1.19 (Figs. 1a and S1c).
2.2.2. Synthesis of PDEA36-ss-PEO104-ss-PDEA36 triblock copolymer (Scheme 3)In a typical run, DEA (5.08 g, 27.42 mmol), CPDB (33.2 mg, 0.15 mmol), and AIBN (4.9 mg, 0.03 mmol) were dissolved in THF (3 mL) with a magnetic stirring bar. The tube was carefully degassed by three freeze-pump-thaw cycles and then sealed under vacuum. After being thermostated at 70 ℃ in an oil bath and stirred for 16 h, the reaction tube was quenched into liquid nitrogen and opened, and the reaction mixture was then precipitated into an excess of petroleum ether. The above dissolution-precipitation cycle was repeated three times. PDEA was obtained as a pale red solid (4.1 g, yield: 80%). The molecular weight and molecular weight distribution of PDEA homopolymer were determined by GPC using THF as the eluent, revealing an Mn of 10.5 kDa and Mw/Mn of 1.06 (Fig. 1b). The actual degree of polymerization (DP) of the PDEA homopolymer was determined to be 36 by 1H NMR analysis recorded in CDCl3 and the obtained homopolymer was denoted as PDEA36 (Fig. S2a).
In the second step, the HO-PEO104-OH (9.0 g, 1.8 mmol) was dissolved in 30 mL dry toluene, and then succinic anhydride (1.0 g, 10 mmol) was added. After the mixture was stirred at 60 ℃ for 20 h, toluene was removed, and the residue was dissolved in CH2Cl2. Then, the solution was precipitated into an excess of diethyl ether. The above dissolution-precipitation cycle was repeated three times, and the obtained product was dried in a vacuum oven at 40 ℃. Then PEO with two terminal carboxyl groups (HOOC-PEO104-COOH) was obtained (7 g, yield: 76%).
HOOC-PEO104-COOH (1.1 g, 0.22 mmol), PyDS-OH (93.6 mg, 0.5 mmol), DCC (103 mg, 0.5 mmol), and DMAP (6 mg, 0.05 mmol) were dissolved in 40 mL dry CH2Cl2, and the reaction mixture was stirred at room temperature for 48 h. After removing insoluble salts by filtration, the filtrates were concentrated on a rotary evaporator and then precipitated into an excess of diethyl ether. The above dissolution-precipitation cycle was repeated three times. After drying in a vacuum oven at 40 ℃, the resulting PyDS-PEO104-PyDS was obtained.
At last, PDEA36 (500 mg, 0.075 mmol) and n-butylamine (16 mg, 0.225 mmol) were dissolved in THF. The reaction proceeded under nitrogen for 2 h until the solution color changed from light red to yellow. After the solvent and excess n-butylamine were removed under nitrogen, pre-degassed PyDS-PEO104-PyDS (133 mg, 0.025 mmol) in THF and 20 mL of glacial acetic acid were added and the reaction mixture was stirred at room temperature under nitrogen for another 24 h. The resulting polymer was precipitated into an excess of diethyl ether to give target triblock copolymer PDEA36-ss-PEO104-ss-PDEA36 (210 mg, 45%). The molecular weight and molecular weight distribution of PDEA36-ss-PEO104-ss-PDEA36 were determined by GPC using THF as the eluent, revealing an Mn of 37.1 kDa and Mw/Mn of 1.23 (Figs. 1b and S2c).
2.3. Preparation of triblock copolymer micellesIn a typical self-assembly procedure, 50 mg PMMA36-ss-PMEO3MA90-ss-PMMA36 triblock copolymer was dissolved in 5 mL THF, stirring and maintained at a constant temperature in a water bath for 20 min. Then, 3 mL water was slowly added dropwise through a syringe at a rate of 1 drop every 5 s under stirring. After that, the mixture was left stirring for an additional 12 h. THF was removed by dialysis (MWCO 3.5 kDa) against deionized water for 3 days.
The triblock copolymer of PDEA36-ss-PEO104-ss-PDEA36 was directly dissolved in deionized water at pH 3 with a concentration of 1.0 g/L. The formation of micellar nanoparticles was induced by adjusting the solution to pH 9 with 0.1 mol/L NaOH aqueous solution.
2.4. Preparation of gold nanoparticle incorporated hybrid triblock copolymer micellesThe colloidal gold nanoparticles was prepared by reduction of chloroauric acid (HAuCl4) with THPC, hereinafter termed as THPC-stabilized gold nanoparticles (~2 nm in diameter), where the method described by Pham et al. [21] was adopted. In a typical run, 0.5 mL aliquot of 1 mol/L NaOH and 1 mL THPC solution (prepared by adding 12 mL THPC (80%) in water to 1 mL HPLC-grade water) were added to 45 mL HPLC water. The reaction mixture was stirred for 5 min, and 10 mL HAuCl4 (5 mmol/L) solutionwas added rapidly. The color changed from yellow to dark brown, indicating the formation of small (~2 nm) THPC-stabilized gold nanoparticles. Then, 30 mL THPC-stabilized gold nanoparticle solution was added to 5 mL micellar dispersion. After stirring at room temperature for 12 h, the gold nanoparticle incorporated hybrid micelles were purified via centrifugation.
3. Results and discussion 3.1. Synthesis of cleavable ABA triblock copolymers PMMA36-ss-PMEO3MA90-ss-PMMA36 and PDEA36-ss-PEO104-ss-PDEA36It is highly desirable to employ an appropriate copolymer micellar template possessing stronger Au-binding ligands for the covalent assembly of AuNPs. Among all the synthesized block copolymers, cleavable block copolymers with the ability to be cleaved under specific external stimulus should be given a great deal of emphasis for their most probably ability of installing AuNPs via covalent interaction at the cleavage site [22, 23]. Yang et al. synthesized polystyrene-b-poly(n-butyl acrylate) with a thermocleavable initiator. When samples were heated to 120 ℃ in the presence of phenylhydrazine, the block copolymers were cleaved into two homopolymer blocks [24]. Russell et al. reported polystyrene-b-poly(methyl methacrylate) copolymers containing [4π + 4π] anthracene photodimer at the junction point between the two blocks, and this diblock copolymer could be cleaved to the parent homopolymer blocks upon heating above 135 ℃ [25]. The synthetic strategy reported by Thayumanavan et al. draw our attention. They synthesized cleavable AB diblock copolymer from two homopolymers obtained by ATRP with an initiator containing pyridyldisulfide moiety via substitution reaction between pyridyldisulfide groups and thiol groups [18]. The disulfide functionality between the two blocks could be cleaved using a reducing agent under mild condition, and gold nanoparticles could be facilely introduced into the nanostructures fabricated from the disulfide group-rich copolymers due to strong interaction between Au-S bonds [26-34].
In our studies, the target ABA triblock copolymer, PMMA36-ss-PMEO3MA90-ss-PMMA36 was synthesized via the substitution reaction of PyDS-PMMA and HS-PMEO3MA90-SH homopolymers (Scheme 2). Specifically, PyDS-PMMA was firstly synthesized according to procedures reported by Thayumanavan et al. [18] via ATRP. Fig. S1a shows the 1H NMR spectrum of the synthesized PyDS-PMMA with the relevant signals labeled. It could be clearly seen that all signals were visible. The signals at d = 1.8, 3.0-4.3, and 7-80.5 ppm were ascribed to methyl protons (g), methylene group (e, f), and pyridyl group (a-d) derived from the ATRP initiator of PyDS-Br, respectively. The characteristic signal at d = 0.9-3.6 and 1.8 were ascribed to methyl protons (i, j) and methylene group (h) of MMA, respectively. Based on the integral ratio of peaks j and f the degree of polymerization, DP, of MMA was calculated to be 36. Then PMEO3MA was synthesized via RAFT polymerization. 1H NMR spectrum of PMEO3MA was shown in Fig. S1b. The resonance signals at 3.3-4.1 ppm attributed to methylene (a, b) and methyl (c) protons of MEO3MA, indicating the successful polymerization of MEO3MA. Based on the integral ratio of peaks c and d (related to the phenyl protons in RAFT agent 1), the degree of polymerization, DP, of MEO3MA was calculated to be 90, and the dithioester ended PMEO3MA90 was reduced by n-butylamine in THF to obtain HSPMEO3MA90-SH. Finally, in Fig. S1c, it can be clearly seen that all signals characteristic of PMEO3MA and PMMA blocks are visible indicating the successfully substitution reaction of PyDS-PMMA36 and HS-PMEO3MA90-SH homopolymers. The excess PyDS-PMMA36 could be removed easily by precipitation in diethyl ether and the target triblock copolymer, PMMA36-ss-PMEO3MA90-ss-PMMA36, was obtained.
GPC analysis results of PyDS-MMA36, PMEO3MA90 homopolymers, and triblock copolymer PMMA36-ss-PMEO3MA90-ss-PMMA36 were also recorded (Fig. 1a). It reveal mono-modal and symmetric peaks with an Mn of ~6500 and ~18, 500, and a polydispersity, Mw/Mn, of 1.07 and 1.13, for PyDS-MMA and PMEO3MA, respectively. The elution peak of the triblock copolymer was shifted toward a higher molecular weight compared to the two homopolymer precursors, suggesting that the target triblock copolymer had been achieved. Moreover, the elution peak of triblock copolymer was also mono-modal and relatively symmetric and did not show any discernible tailing at the lower molecular weight side, confirming nearly no homopolymer residue in the triblock copolymer. The molecular weight and molecular weight distribution of triblock copolymer was characterized by GPC analysis in THF: Mn =, Mw/Mn = 1.19.
PDEA36-ss-PEO104-ss-PDEA36 was synthesized via the substitution reaction of PDEA-SH and PyDS-PEO-PyDS homopolymers (Scheme 3). Specifically, PDEA was firstly synthesized via RAFT polymerization. All signals attributed to DEA were visible in 1H NMR spectrum and the relevant signals were labeled (Fig. S2a). For example the characteristic signals at d = 0.9-1.0 and 1.8-4.0 ppm were ascribed to methyl protons (a, f) and methylene group (b-e) of DEA, respectively. Based on the integral ratio of peaks c and g (related to the phenyl protons in RAFT agent), the degree of polymerization, DP, of PDEA was calculated to be 36. After reduction of the dithioester end of PDEA36 by n-butylamine in THF, PDEA36-SH was obtained. Then PyDS-PEO104-PyDS was prepared by the esterification reaction of HOOC-PEO-COOH with PyDS-OH in the presence of DCC and DMAP (Scheme 3). 1H NMR spectroscopy studies indicated that the esterification reaction was essentially completed (Fig. S2b). The signal at 3.7 ppm (peak a) could be ascribed to the methylene protons of PEO main chain, whereas signal at 8.5 ppm (peak b) was ascribed to pyridine proton of terminal PyDS-OH group. By comparing integral ratios of peak a to that of b, the degree of end group functionalization was calculated to be nearly 100%. Finally, PDEA36-ss-PEO104-ss-PDEA36 was synthesized via the substitution reaction of PDEA36-SH and PyDS-PEO104-PyDS, and the excess PDEA36-SH could be removed easily by precipitation in diethyl ether. 1H NMR spectrum in Fig. S2c showed that all characteristic signals of PEO and PDEA blocks were visible.
GPC analysis results of PyDS-PEO104-PyDS, PDEA36-SH homopolymers, and triblock copolymer PDEA36-ss-PEO104-ss-PDEA36 were also recorded (Fig. 1b). It reveal mono-modal and symmetric peaks with an Mn of ~ and ~, and a polydispersity, Mw/Mn, of 1.09 and 1.08, for PDEA-SH and PyDS-PEO-PyDS homopolymers, respectively. Similar to PMMA36-ss-PMEO3MA90-ss-PMMA36, GPC traces of triblock copolymer PDEA36-ss-PEO104-ss-PDEA36 was shifted toward a higher molecular weight compared to that of the two homopolymers, and the elution peak was also mono-modal and symmetric. The molecular weight and molecular weight distribution of triblock copolymer was characterized by GPC analysis in THF: Mn =, Mw/Mn = 1.23. Thus the two reductioncleavable triblock copolymers were successfully synthesized.
To determine the reduction-cleavability of the triblock copolymer, GPC traces of the triblock copolymers were analyzed before and after mixing with excess dithiothreitol (DTT). The disulfide moieties in triblock copolymers were reduced to thiol group in the presence of DTT, resulting triblock copolymer cleavage to mixtures of homopolymers. As shown in Fig. 1c, d, the GPC traces of triblock copolymers were shifted to a lower molecular weight and divided to two peaks corresponding to the elution time peak of the two homopolymer precursors after an excess DTT treatment, indicating the cleavability of the triblock copolymer under reduction microenvironment.
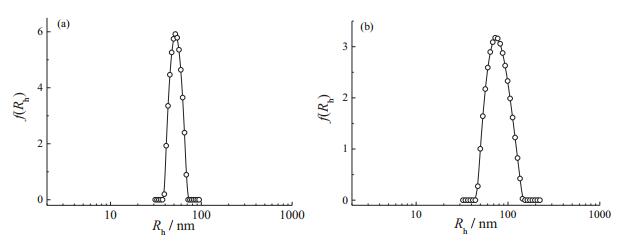
3.2. Micellization of ABA triblock copolymersThe micellization behavior of amphiphilic [35-39] and double hydrophilic block copolymer (DHBCs) [40, 41] have been widely investigated. The two ABA triblock copolymers used here were amphiphilic and DHBCs, respectively. DLS was employed to characterize the micellization behavior of these triblock copolymers. Fig. 2 shows the typical hydrodynamic radius distributions, f (Rh), of the micellar dispersion of PMMA36-ss-PMEO3MA90-ss-PMMA36 and PDEA36-ss-PEO104-ss-PDEA36. For PMMA36-ss-PMEO3MA90-ss-PMMA36, DLS revealed an intensity-average hydrodynamic radius, f (Rh), of 52 nm and a relatively narrow polydisperdity, μ2/Γ2, of 0.02 (Fig. 2a). And the micelles fabricated from PDEA36-ss-PEO104-ss-PDEA36, exhibited a polydispersity, μ2/Γ2, of 0.04 and a hydrodynamic radius, f (Rh), of 78 nm (Fig. 2b).

|
Download:
|
| Fig. 2. Typical hydrodynamic radius distributions, f (Rh), obtained for aqueous micellar dispersions (1.0 g/L, 25 ℃) of (a) PMMA36-ss-PMEO3MA90-ss-PMMA36 (pH 7) and (b) PDEA36-ss-PEO104-ss-PDEA36 (pH 9). | |
{kind=link}
The micellization of the ABA triblock copolymer was further confirmed by TEM observations. Fig. 3 shows the typical TEM images of micelles fabricated from PMMA36-ss-PMEO3MA90-ss-PMMA36 (Fig. 3a) and PDEA36-ss-PEO104-ss-PDEA36 (Fig. 3b), respectively. The morphology and the size distributions of the micelles were spherical and relatively narrow in both cases. The particle sizes determined by TEM were in the range of 50-60 nm and 70-80 nm in diameter, for Fig. 3a and b, respectively, which were smaller than the particle sizes obtained by DLS for the same micellar dispersions. For block copolymer micelles, it had been well-established that particle size determined by TEM were typically smaller than those by DLS because the former reflected conformations in dry state [42].

|
Download:
|
| Fig. 3. TEM images of micelles self-assembly from (a) PMMA36-ss-PMEO3MA90-ss-PMMA36 (pH 7) and (b) PDEA36-ss-PEO104-ss-PDEA36 (pH 9). | |
{kind=link}
3.3. Cleavability of the ABA triblock copolymers
The two ABA triblock copolymer were formed by disulfide bond connection. As we known, the disulfide bonds can be readily cleaved using a reducing agent such as DTT. And these were demonstrated by measuring the GPC traces of the triblock copolymers before and after adding DTT (Fig. 1c, d). Compared to the traces of non-added DTT samples, we can conclude that the ABA triblock copolymers were almost completed cleaved by DTT. It clearly demonstrated the versatility of our strategy.
Since we have demonstrated that the ABA triblock copolymers can be cleaved by reducing agent, the micelles fabricated by these polymers should also can be cleaved by reducing the disulfide bonds. Considering the morphology of the micellar structure, the disulfide bonds were located at the interface of the micellar core and shell. If the disulfide bonds were reduced to thiol group, the core and shell separated, and the insoluble core would be precipitated. As the digital photograph shown in Fig. 4, upon adding reducing agent sodium borohydride (NaBH4) to the bluish PMMA36-ss-PMEO3MA90-ss-PMMA36 micellar dispersion, the solution quickly appeared turbidity which indicated the macroscopic phase separation of the solution due to the hydrophilic PMEO3MA corona was cleaved.

|
Download:
|
| Fig. 4. Digital photographs obtained for the aqueous micellar solution (1.0 g/L, 25 ℃) of PMMA36-ss-PMEO3MA90-ss-PMMA36 before and after the addition of NaBH4 or THPC-stabilized gold nanoparticles, respectively. | |
{kind=link}
3.4. Preparation of gold nanoparticle incorporated hybrid micelles
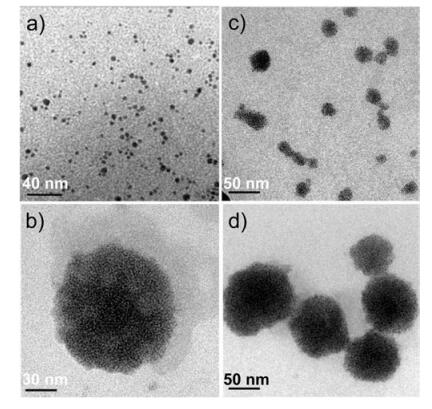
Note that Au-S bond was a strong chemical bond, we then attempted to take the disulfide bonds in the interface of micellar core and shell reacting with weak-interaction stabilized gold nanoparticles to fabricate AuNPs incorporated hybrid micelles. We have shown that the particle sizes prepared by the two ABA triblock copolymers were about in the range of 50-80 nm in diameter. Considering the steric hindrance in the micellar core/ shell interface affecting the diffusion process of nanoparticles, THPC-stabilized gold nanoparticles with relatively smaller diameter of ~2 nm were synthesized according to previous literature reports [21] (Fig. 5a).

|
Download:
|
| Fig. 5. TEM images of (a) THPC-stabilized gold nanoparticles and gold nanoparticles incorporated hybrid micelles prepared from (b-c) PMMA36-ss-PMEO3MA90-ss-PMMA36 and (d) PDEA36-ss-PEO104-ss-PDEA36. | |
{kind=link}
After adding the THPC-stabilized gold nanoparticles to PMMA36-ss-PMEO3MA90-ss-PMMA36 micellar dispersion, we obtained a homogeneous darkness solution (Fig. 4). DLS was then employed to characterize the micellar sizes and size distributions before and after adding gold nanoparticles. As shown in Fig. S3, the intensity-average hydrodynamic radius, f (Rh), did not changed significantly before and after adding THPC-stabilized gold nanoparticles. Although the size distribution of the solution in the presence of gold nanoparticles was a little broader than that in the absence of gold nanoparticles, we could conclude that the adding gold nanoparticles did not significantly affect the nanostructures of the copolymer micelles.
TEM was further used to observe the hybrid micellar structures. From the TEM images of gold nanoparticle incorporated hybrid micelles of PMMA36-ss-PMEO3MA90-ss-PMMA36 (Fig. 5b, c) and PDEA36-ss-PEO104-ss-PDEA36 (Fig. 5d), we could clearly observe that the gold nanoparticles homogeneous distributed in the outer surface of the micellar core. Because S-Au bonds were more stable than the disulfide bonds and the weak interaction between gold nanoparticles and THPC-stabilized gold nanoparticles could be easily inserted into the interface of micellar core and shell to form an inner gold nanoshell. And we believe that the hydrophilic block was still in the outer shell of the hybrid micelle. Temperature dependence of optical transmittance of gold nanoparticle incorporated hybrid micelles fabricated from PMMA36-ss-PMEO3MA90-ss-PMMA36 was measured, and the transmittance of the hybrid micellar dispersion was decreased when the temperature was higher than 60 ℃ (Fig. S4), which was close to the lower critical solution temperature (LCST) of PMEO3MA [43], indicating PMEO3MA was still at the particle surface after incorporation of gold nanoparticles. It should be noted that we found gold nanoparticles incorporated PMMA36-ss-PMEO3MA90-ss-PMMA36 micelles with diameters of ~50-60 nm in Fig. 5b, and also found those with the diameters smaller than 50-60 nm shown in Fig. 5c, which is most probably due to the border size distribution after adding gold nanoparticles.
4. ConclusionThe present study demonstrated the successful syntheses of two cleavable ABA triblock copolymers, PMMA-ss-PMEO3MA-ss-PMMA and PDEA-ss-PEO-ss-PDEA via a facile substitution reaction from homopolymer precursors. Supramolecular self-assembly of these two triblock copolymers in aqueous solutions induced the formation of spherical micelles. It is most interesting that the gold nanoparticles can precisely incorporated at the core/shell interface of micelles, and these reduction-responsive hybrid nanospheres with novel structure should have potential applications, such as disease diagnosis and controlled drug delivery.
AcknowledgmentsThe financial support from the [21_TD$DIFF][1_TD$DIFF]National Natural Scientific Foundation of China (NNSFC) Project (Nos. 51690150, 51690154, and 21674103) and Anhui Provincial Natural Scientific Foundation project (No. 1508085QB43) is gratefully acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.03.020.
| [1] | M.C. Daniel, D. Astruc. Gold nanoparticles:assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology catalysis, and nanotechnology. Chem. Rev. 104(2004)293–346. DOI:10.1021/cr030698+ |
| [2] | J. Shan, H. Tenhu. Recent advances in polymer protected gold nanoparticles:synthesis, properties and applications. Chem. Commun. (2007)4580–4598. |
| [3] | J.M. Tam, J.O. Tam, A. Murthy, D.R. Ingram, L.L. Ma, K. Travis, K.P. Johnston, K.V. Sokolov. Controlled assembly of biodegradable plasmonic nanoclusters for near-infrared imaging and therapeutic applications. ACS Nano 4(2010)2178–2184. DOI:10.1021/nn9015746 |
| [4] | Y.F. Huang, K. Xia, N.Y. He, Z.X. Lu, L.M. Zhang, Y. Deng, L.B. Nie. Size-tunable synthesis of gold nanorods using pyrogallol as a reducing agent. Sci. China Chem. 58(2015)1759–1765. DOI:10.1007/s11426-015-5437-3 |
| [5] | X.H. Zu, Z.H. Jian, G.B. Yi, H.L. Huang, B.B. Zhong, H.S. Luo, J.R. Huang, C. Wang. Surface-enhanced Raman scattering effect of ordered gold nanoparticle array for rhodamine B with different morphologies. Chin. J. Polym. Sci. 33(2015)1470–1476. DOI:10.1007/s10118-015-1690-3 |
| [6] | M.H. Mashhadizadeh, R.P. Talemi. Application of diazo-thiourea and gold nano-particles in the design of a highly sensitive and selective DNA biosensor. Chin. Chem. Lett. 26(2015)160–166. DOI:10.1016/j.cclet.2014.09.004 |
| [7] | M. Filali, M.A.R. Meier, U.S. Schubert, J.F. Gohy. Star-block copolymers as templates for the preparation of stable gold nanoparticles. Langmuir 21(2005)7995–8000. DOI:10.1021/la050377o |
| [8] | J.B. Li, L.Q. Shi, Y.L. An, Y. Li, X. Chen, H.J. Dong. Reverse micelles of star-block copolymer as nanoreactors for preparation of gold nanoparticles. Polymer 47(2006)8480–8487. DOI:10.1016/j.polymer.2006.09.071 |
| [9] | F. Zhao, L. Li, Y.C. Tian, J.J. Liu, J.J. Wang, Z.M. Zhou, C.X. Lv, X.H. Guo. Preparation of Au/Ag multilayers via layer-by-layer self-assembly in spherical polyelectrolyte brushes and their catalytic activity. Chin. J. Polym. Sci. 33(2015)1421–1430. DOI:10.1007/s10118-015-1700-5 |
| [10] | T. Sakai, P. Alexandridis. Size-and shape-controlled synthesis of colloidal gold through autoreduction of the auric cation by poly(ethylene oxide)-poly (propylene oxide) block copolymers in aqueous solutions at ambient conditions. Nanotechnology 14(2005)S344–S353. |
| [11] | T. Sakai, P. Alexandridis. Ag and au monometallic and bimetallic colloids:morphogenesis in amphiphilic block copolymer solutions. Chem. Mater. 18(2006)2577–2583. DOI:10.1021/cm051757y |
| [12] | X. Chen, Y. Liu, Y. An, J. Lu, J. Li, D. Xiong, L. Shi. Novel structured composites formed from gold nanopartnocles and diblock copolymers. Macromol. Rapid Commun. 28(2007)1350–1355. DOI:10.1002/(ISSN)1521-3927 |
| [13] | H.X. Xu, J. Xu, X.Z. Jiang, Z.Y. Zhu, J.Y. Rao, J. Yin, T. Wu, H.W. Liu, S.Y. Liu. Thermosensitive unimolecular micelles surface-decorated with gold nanoparticles of tunable spatial distribution. Chem. Mater. 19(2007)2489–2494. DOI:10.1021/cm070088g |
| [14] | W.R. Lee, M.G. Kim, J.R. Choi, J.I. Park, S.J. Ko, S.J. Oh, J. Cheon. Redox-transmetalation process as a generalized synthetic strategy for core-shell magnetic nanoparticles. J. Am. Chem. Soc. 127(2005)16090–16097. DOI:10.1021/ja053659j |
| [15] | W.R. Zhao, J.L. Gu, L.X. Zhang, H.R. Chen, J.L. Shi. Fabrication of uniform magnetic nanocomposite spheres with a magnetic core/mesoporous silica shell structure. J. Am. Chem. Soc. 127(2005)8916–8917. DOI:10.1021/ja051113r |
| [16] | L.Y. Wang, J. Luo, Q. Fan, M. Suzuki, I.S. Suzuki, M.H. Engelhard, Y.H. Lin, N. Kim, J.Q. Wang, C.J. Zhong. Monodispersed core-shell Fe3O4@Au nanoparticles. J. Phys. Chem. B 109(2005)21593–21601. DOI:10.1021/jp0543429 |
| [17] | H. Kobayashi, M. Yamauchi, H. Kitagawa, Y. Kubota, K. Kato, M. Takata. Hydrogen absorption in the core/shell interface of Pd/Pt nanoparticles. J. Am. Chem. Soc. 130(2008)1818. DOI:10.1021/ja078126k |
| [18] | A. Klaikherd, S. Ghosh, S. Thayumanavan. A facile method for the synthesis of cleavable block copolymers from ATRP-based homopolymers. Macromolecules 40(2007)8518–8520. DOI:10.1021/ma071852n |
| [19] | K. Satoh, D.H. Lee, K. Nagai, M. Kamigaito. Precision synthesis of bio-based acrylic thermoplastic elastomer by RAFT polymerization of itaconic acid derivatives. Macromol. Rapid Commun. 35(2014)161–167. DOI:10.1002/marc.201300638 |
| [20] | H. Zhang, X. Tong, Y. Zhao. Diverse thermoresponsive behaviors of uncharged UCST block copolymer micelles in physiological medium. Langmuir 30(2014)11433–11441. DOI:10.1021/la5026334 |
| [21] | T. Pham, J.B. Jackson, N.J. Halas, T.R. Lee. Preparation and characterization of gold nanoshells coated with self-assembled monolayers. Langmuir 18(2002)4915–4920. DOI:10.1021/la015561y |
| [22] | Z. Ge, S. Liu. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem. Soc. Rev. 42(2013)7289–7325. DOI:10.1039/c3cs60048c |
| [23] | H.H. Liu, D.D. Tang, R.P. Tang, Y.L. Zhao. Synthesis of multifunctional ABC stars with a reduction-labile arm by consecutive ROP, RAFT and ATRP processes. Sci. China Chem. 58(2015)1724–1733. DOI:10.1007/s11426-015-5436-4 |
| [24] | W. Tang, J. He, Y. Yang. A facile synthesis of cleavable block copolymers via tandem polymerizations of NMRP and ATRP. J. Macromol. Sci. A 43(2006)1553–1567. DOI:10.1080/10601320600896850 |
| [25] | J.T. Goldbach, T.P. Russell, J. Penelle. Synthesis and thin film characterization of poly(styrene-block-methyl methacrylate) containing an anthracene dimer photocleavable junction point. Macromolecules 35(2002)4271–4276. DOI:10.1021/ma011940m |
| [26] | M.H. Lee, Z. Yang, C.W. Lim, Y.H. Lee, S. Dongbang, C. Kang, J.S. Kim. Disulfidecleavage-triggered chemosensors and their biological applications. Chem. Rev. 113(2013)5071–5109. DOI:10.1021/cr300358b |
| [27] | Y. Jiang, G. Liu, X. Wang, J. Hu, G. Zhang, S. Liu. Cytosol-specific fluorogenic reactions for visualizing intracellular disintegration of responsive polymeric nanocarriers and triggered drug release. Macromolecules 48(2015)764–774. DOI:10.1021/ma502389w |
| [28] | J. Hu, X. Wang, Y. Qian, Y. Yu, Y. Jiang, G. Zhang, S. Liu. Cytoplasmic reactive cationic amphiphiles for efficient intracellular delivery and self-reporting smart release. Macromolecules 48(2015)5959–5968. DOI:10.1021/acs.macromol.5b01110 |
| [29] | X. Hu, G. Liu, Y. Li, X. Wang, S. Liu. Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals. J. Am. Chem. Soc. 137(2015)362–368. DOI:10.1021/ja5105848 |
| [30] | J. Song, L. Cheng, A. Liu, J. Yin, M. Kuang, H. Duan. Plasmonic vesicles of amphiphilic gold nanocrystals:self-assembly and external-stimuli-triggered destruction. J. Am. Chem. Soc. 133(2011)10760–10763. DOI:10.1021/ja204387w |
| [31] | J. Hu, T. Wu, G. Zhang, S. Liu. Efficient synthesis of single gold nanoparticle hybrid amphiphilic triblock copolymers and their controlled self-assembly. J. Am. Chem. Soc. 134(2012)7624–7627. DOI:10.1021/ja302019q |
| [32] | Y. Mai, L. Xiao, A. Eisenberg. Morphological control in aggregates of amphiphilic cylindrical metal-polymer brushes. Macromolecules 46(2013)3183–3189. DOI:10.1021/ma400236g |
| [33] | H. Deng, Y. Zhong, M. Du, Q. Liu, Z. Fan, F. Dai, X. Zhang. Theranostic selfassembly structure of gold nanoparticles for NIR photothermal therapy and XRay computed tomography imaging. Theranostics 4(2014)904–918. DOI:10.7150/thno.9448 |
| [34] | S.L. Yi, M.C. Li, X.Q. Hu, W.M. Mo, Z.L. Shen. An efficient and convenient method for the preparation of disulfides from thiols using oxygen as oxidant catalyzed by tert-butyl nitrite. Chin. Chem. Lett. 27(2016)1505–1508. DOI:10.1016/j.cclet.2016.03.016 |
| [35] | Y. Mai, A. Eisenberg. Self-assembly of block copolymers. Chem. Soc. Rev. 41(2012)5969–5985. DOI:10.1039/c2cs35115c |
| [36] | J. Chu, Q.L. Lv, C.L. Guo, D.Z. Xu, K. Wang, M.Y. Liu, H.Y. Huang, X.Y. Zhang, Y. Wei. One-step preparation of branched PEG functionalized AIE-active luminescent polymeric nanoprobes. Sci. China Chem. 59(2016)1003–1009. DOI:10.1007/s11426-016-5578-z |
| [37] | C.Y. Wang, Q. Yuan, S.G. Yang, J. Xu. Effect of water content on the size and membrane thickness of polystyrene-block-poly(ethylene oxide) vesicles. Chin. J. Polym. Sci. 33(2015)661–668. DOI:10.1007/s10118-015-1618-y |
| [38] | J.C. Chen, J.Z. Li, J.H. Liu, L.Q. Xu. Amphiphilic poly(ethylene glycol)-b-poly (ethylene brassylate) copolymers:one-pot synthesis, self-assembly, and controlled drug release. Chin. Chem. Lett. 26(2015)1319–1321. DOI:10.1016/j.cclet.2015.05.050 |
| [39] | R.B. Wei, X.G. Wang, Y.N. He. Synthesis, self-assembly and photo-responsive behavior of AB(2) shaped amphiphilic azo block copolymer. Chin. Chem. Lett. 26(2015)857–861. DOI:10.1016/j.cclet.2015.04.019 |
| [40] | Z. Ge, D. Xie, D. Chen, X. Jiang, Y. Zhang, H. Liu, S. Liu. Stimuli-Responsive double hydrophilic block copolymer micelles with switchable catalytic activity. Macromolecules 40(2007)3538–3546. DOI:10.1021/ma070550i |
| [41] | J.J. Xiao, X.B. Li, X. Wang, C.W. Yi, S.P. Su. Effect of temperature-responsive solution behavior of PNIPAM-b-PPEOMA-b-PNIPAM on its inclusion complexation with alpha-cyclodextrin. Chin. J. Polym. Sci. 33(2015)456–464. DOI:10.1007/s10118-015-1598-y |
| [42] | H. Huang, E.E. Remsen, T. Kowalewski, K.L. Wooley. Nanocages derived from shell cross-linked micelle templates. J. Am. Chem. Soc. 121(1999)3805–3806. DOI:10.1021/ja983610w |
| [43] | J.-F. Lutz. Polymerization of oligo(ethylene glycol) (meth)acrylates:toward newgenerations of smart biocompatible materials. J. Polym. Sci. Part A:Polym. Chem. 46(2008)3459–3470. DOI:10.1002/(ISSN)1099-0518 |