 2017, Vol. 28
2017, Vol. 28 



b Shanghai Key Laboratory of Magnetic Resonance, Institute of Functional Materials, School of Physics and Materials Science, East China Normal University, Shanghai 200062, China
For drugs to be delivered as a solid, the selection of their specific solid forms plays a central role in optimizing the thermal stability, solubility, dissolution rate, and consequently the bioavailability of these drugs [1]. With the development of combinatorial chemistry and high-throughput screening in drug discovery, more and more drug candidates possess challenging dissolution profiles, such as low solubility and dissolution rate [2-4]. A number of solid forms has been exploited to address this key issue [5], including the metastable polymorphs of the pure drug molecule [6, 7], drugcoformer cocrystals [8-11], salts [12, 13] and drug/polymer solid dispersions [14, 15]. Although these approaches have been successful, the study of new solid forms is still needed to provide more options to achieve the desirable pharmaceutical profiles of drugs.
In addition to the aforementioned solid forms, recently certain drug molecules were found to form a new solid form together with multiple types of linear polymers, i.e., the crystalline inclusion complexes (ICs) [16-18]. In a drug-polymer IC, the drug molecules form a crystal lattice framework with parallel, isolated channels accommodating polymer chains of extended conformations. It seems that the guest polymers mainly act as void fillers to stabilize the channel structure of the host lattice. Therefore, isostructural ICs can be readily formed between the same drug molecule and different guest polymers with suitable chain cross-section sizes. As a relatively new pharmaceutical solid form, the generality and the structure-property relationship of the drug/polymer ICs are yet to be understood.
In the previous publication, we reported the dissolution behavior and thermal stability of several drug ICs formed with homopolymers [19, 20]. It was found that, the apparent solubility and dissolution rate of the poorly water-soluble drug griseofulvin were significantly enhanced by using hydrophilic poly(ethylene glycol) (PEG) as the guest polymer [19]. In contrast, the ICs formed by the drug diflunisal (DIF) and hydrophobic poly(ε-caprolactone) (PCL) exhibited decreased solubility and dissolution rate compared with the pure DIF crystals [20]. The thermal stability of these drug/ polymer ICs also showed a strong dependence on both the type and chain length of the guest polymers, indicating the potential of drug-polymer ICs as a new approach to modulate the pharmaceutical profiles of drugs.
In comparison to the homopolymers used in the previous studies, here we extend the drug/polymer ICs to block copolymers, and study the influence of block copolymers on the formation, thermal stability and dissolution behavior of drug/polymer ICs. In our previous work, PCL was found to readily form ICs with DIF [16]. However, even after extensive experiments, we could not obtain DIF/PEG ICs probably due to its low packing efficiency in the IC channels. Interestingly, in this current study where the diblock copolymers of PEG and PCL were used as the guest, the PEG block can indeed be included in the DIF ICs when the PCL block is long enough. In addition, we found that the block length of the guest copolymers had significant influence on the thermal stability, aqueous solubility, as well as dissolution rate of DIF/PEG-b-PCL ICs. It should be noted that, outside the pharmaceutical field urea ICs and cyclodextrin ICs formed with block copolymers were extensively studied and showed block-selective inclusion complexation [21-26]. The different blocks of a copolymer may or may not be included in the same IC channels, which is mainly determined by the fit between the chain cross-section sizes and the channel dimension [21-25]. However, in certain ICs even though the blocks can be included individually, simultaneous inclusion of these blocks was not observed when the copolymer was used [26].
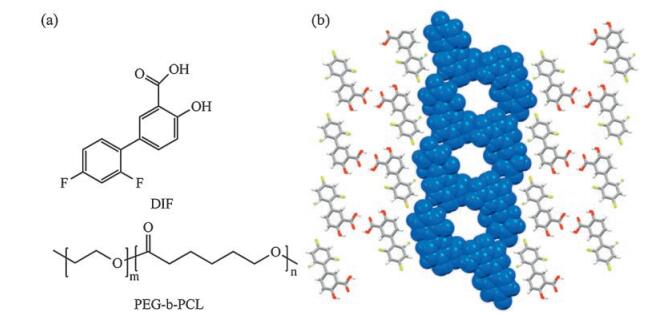
The molecular formulas of DIF and PEG-b-PCL, as well as the channel structure of DIF/PCL IC [16], are shown in Fig. 1. Throughout the current study, the number-averaged molecular weight (Mn) of the PEG block was kept constant at 2k Da, while those of the PCL block varied as 0.5 kDa, 1 kDa, 2 kDa, 3 kDa, 4 kDa, 5 kDa, 8 kDa and 20 kDa. For comparison, DIF crystals obtained with PEG (Mn 2 kDa) and PCL (Mn 10 kDa) homopolymers were also included in the current study.

|
Download:
|
| Fig. 1. (a) Structural formula of DIF and PEG-b-PCL. (b) Crystal structure of DIF/PCL IC with the channel structure highlighted in the space-filling representation [16] | |
2. Results and discussion 2.1. Structural confirmation of DIF/PEG-b-PCL ICs with both blocks in the channels
The cocrystallization between DIF and the guest polymers was carried out from ethyl acetate/heptane solution, and the structural features of the resulting solids were first characterized by PXRD (Fig. 2). It can be seen clearly that, the crystal structures of the resulting solids varied with the block lengths of the guest polymers.

|
Download:
|
| Fig. 2. (a) PXRD patterns of the resulting solids with different guest polymers. Diffraction pattern of DIF Form I is also included for comparison. (b) Magnification of the diffraction patterns to show more clearly the slight shifts of the characteristic peaks to lower 2θ angles for DIF/PEG-b-PCL compared with DIF/PCL. | |
DIF IC crystals were generated with the PCL homopolymer (PCL10k), consistent with that observed in our previous publication [16]. In contrast, when the PEG homopolymer (PEG2k) was used as the guest, the obtained crystals were identified as DIF Form I [27]. The tendency for IC formation increased with the length of the PCL block in the guest copolymers. When the PCL block was PCL0.5k, the resulting solids were found to be mainly the DIF Form I crystals, plus a small amount of the IC crystals (Figs. S1 and S2 in Supporting information). While for DIF/PEG2k-b-PCL1k, a mixture of DIF Form I and the IC crystals was obtained, as evidenced by the coexistence of the characteristic peaks from DIF Form I (e.g., at 4.2°, 13.5°, 14.4°, and 16.6° 2θ) and DIF IC (e.g., at ca. 9.2°, 10.9°, 11.6° and 15.7° 2θ) in the diffraction pattern. When the molecular weight of the PCL block was equal to or larger than 2 kDa, pure IC crystals were obtained. It should be noted that the IC crystal samples contained no excess guest copolymers, as confirmed by the absence of their characteristic diffraction peaks (Figs. 2a and Fig. S3 in Supporting information). It is also worth noting that, though the DIF ICs obtained in this study exhibited highly similar diffraction patterns, slight shifts of the characteristic peaks to lower 2θ angles were observed for DIF/PEG-b-PCL compared with DIF/PCL (Fig. 2b), suggesting the slight expansion of the unit cells in these ICs due to the incorporation of the PEG block in the channel structure.
To confirm the formation of DIF ICs instead of their isostructural solvates [28-30], TGA analysis was performed on the solids obtained in this study (Fig. S4 in Supporting information). No obvious weight loss was observed on the TGA curves before 170 ℃, indicating that no ethyl acetate or heptane solvate formed during the cocrystallization process. At 260 ℃ when almost all DIF was lost, the remaining weight (~10% of the initial weight) of these DIF ICs should be the guest polymers whose obvious thermal decomposition all occurred above 260 ℃ (Fig. S4).
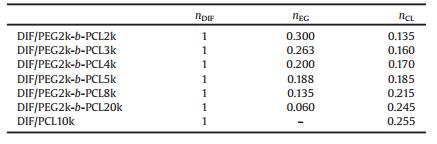
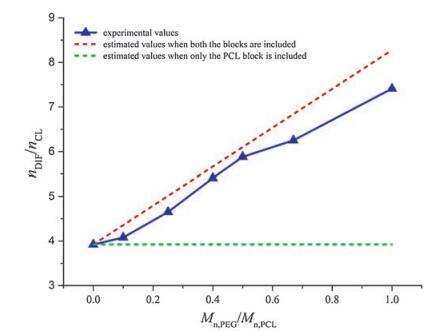
Solution 1H NMR was used to quantify the chemical composition of the DIF ICs (Fig. S5 in Supporting information). The drug: monomer unit molar ratios in the IC crystals are listed in Table 1. The variation of the DIF:CL molar ratios (nDIF/nCL) in the IC crystals with the ratios between the molecular weight of the PEG block and the PCL block (Mn, PEG/Mn, PCL) in the guest polymers is summarized in Fig. 3. If only the PCL block were included in DIF/PEG-b-PCL ICs, their nDIF/nCL would have remained constant (the green dashed line) similar to that of DIF/PCL IC. However, when both the PEG block and the PCL block are included in the IC crystals, the nDIF/nCL alue should vary with Mn, PEG/Mn, PCL. If we assume the PEG block and the PCL block adopt similar chain conformations when they are both included in the IC crystals, one EG unit should be equal to 3/7 CL unit (i.e., 0.429 CL unit) in terms of the chain length along the channel direction. So the relationship nDIF/(0.429nEG + nCL) = 1/ 0.255 should exit in DIF/PEG-b-PCL ICs. At the same time, Mn, PEG/Mn, PCL = 44nEG/114nCL. Combining the above equations gives the relationship between nDIF/nCL and Mn, PEG/Mn, PCL when both the blocks are included: nDIF/nCL = 3.922 + 4.354 Mn, PEG/Mn, PCL (the red dashed line). As clearly shown in Fig. 3, the experimental results were consistent with the second scenario. Therefore, it can be deduced that both the blocks in the guest copolymers are likely to be accommodated in the DIF nanochannels.
|
|
Table 1 Molar ratios of DIF to monomers in the DIF ICs. |

|
Download:
|
| Fig. 3. Variation of the DIF:CL molar ratios in the IC crystals with the ratios between the molecular weights of the PEG block and the PCL block in the guest polymers. Mn, PEG is fixed at 2 kDa in the PEG-b-PCL copolymers. | |
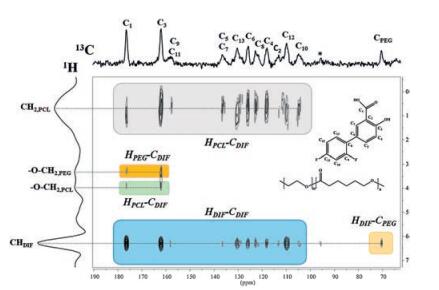
To further confirm the location of the PEG block and the PCL block in the DIF ICs, the FSLG (1H-13C) 2D HETCOR experiment was conducted on DIF/PEG2k-b-PCL2k IC. As shown in Fig. 4, several 1H-13C host-guest correlations are observed at a cross polarization time of 1 ms. The intense cross peaks between the methylene protons of PCL and the aromatic carbons of DIF provide evidence of many strong magnetization interactions between the PCL block and the DIF nanochannels (as indicated by the grey blocks in the Fig. 4). Correlations of the PEG block with DIF molecule are also observable in this spectrum, as confirmed by the polarization transfer from the PEG protons to the DIF carbons, as well as the polarization transfer from the aromatic protons of DIF to the PEG carbons (the yellow blocks). Since the effective polarization transfer indicates the short proton-to-carbon distances [31-33], it can be reasonably concluded that both the PEG block and the PCL block should reside in the nanochannels formed by DIF molecules. In addition, the close contact of the guest copolymers with the DIF nanochannels can also be revealed by the 1H MAS NMR spectra (Fig. S6 in Supporting information). Compared with the line width in the bulk copolymers, significant line broadening was observed for the guest 1H signals in the IC crystals, which is likely caused by the stronger dipolar coupling between the guest protons and the DIF protons [34]. The above results clearly demonstrate that, the PEG block, which could not be included alone, can now be accommodated in the DIF nanochannels associated with the inclusion of the PCL block with enough chain length. Therefore, for guest polymers that can hardly be included alone, block copolymers with IC-forming blocks might provide a general method to trap these polymers into the IC channels.

|
Download:
|
| Fig. 4. 2D 1H-13C HETCOR NMR spectrum of DIF/PEG2k-b-PCL2k IC at a contact time of 1 ms, using FSLG decoupling and 10 kHz spinning speed. The DIF-PCL, DIF-PEG and DIF-DIF cross signals are highlighted in different colors. | |
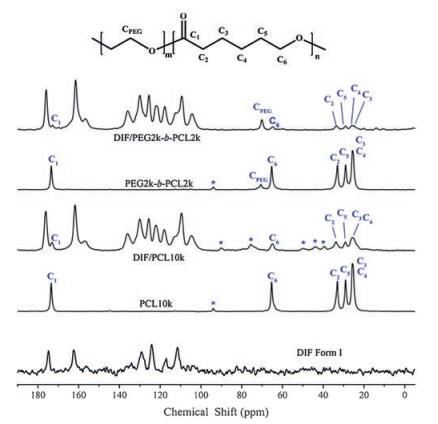
Next, 13C CP/MAS NMR was used to investigate the molecular packing of DIF in the IC crystals and the possible conformations of the PEG and PCL blocks confined in the DIF nanochannels. As shown in Fig. 5, the 13C signals of DIF molecules exhibit close similarities between DIF/PEG2k-b-PCL2k IC and DIF/PCL10k IC, which confirms their isostructural features previously revealed by their PXRD patterns.

|
Download:
|
| Fig. 5. 13C CP/MAS NMR spectra of DIF Form I, DIF ICs and the corresponding guest polymers. The resonances characteristic of the guest polymers can be clearly seen in the DIF ICs. The peaks marked with asterisks indicate spinning sidebands. | |
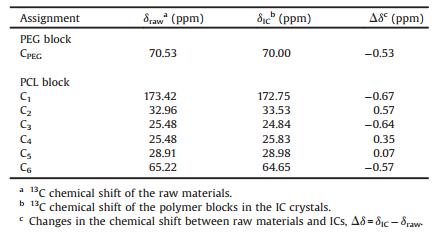
13C signals characteristic of the guest copolymer can be clearly observed in its DIF IC, and exhibit chemical shift changes to various extent compared with those in pure block copolymers. The chemical shift values of the guest copolymer before and after inclusion in the DIF nanochannels are summarized in Table 2. For each carbon of the PEG block and the PCL block, the absolute change of the chemical shift upon inclusion is less than 1 ppm. These small changes in the 13C chemical shift can be mainly attributed to the intermolecular packing effect [21, 35], instead of the conformational changes [36, 37]. Therefore, we propose that the conformations adopted by the PCL block residing in the DIF nanochannels are very similar to the all-trans, planar zigzag conformation in its bulk crystalline material [38], which is similar to those observed in DIF/PCL IC [16] and α-CD/PLLA-b-PCL IC [21]. While for the isolated PEG block, certain helical conformations may be adopted similar to that observed in its crystals [40]. This conclusion is consistent with the aforementioned slight expansion of the unit cells in DIF/PEG-b-PCL ICs, since the PEG block with a helical conformation would possess a larger chain cross-section size compared with the PCL block having a planar zigzag conformation [38-40].
|
|
Table 2 Chemical shift values for PEG2k-b-PCL2k in bulk and in the DIF IC. |
2.2. Thermal stability and dissolution profiles
As illustrated above, DIF can form isostructural ICs with PEG-b-PCL of various block lengths ranging from PCL2k to PCL20k, and it will be interesting to investigate the influence of the block length on the thermal stability and dissolution behavior of the resulting IC crystals.
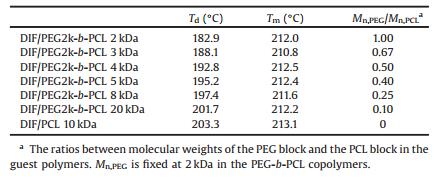
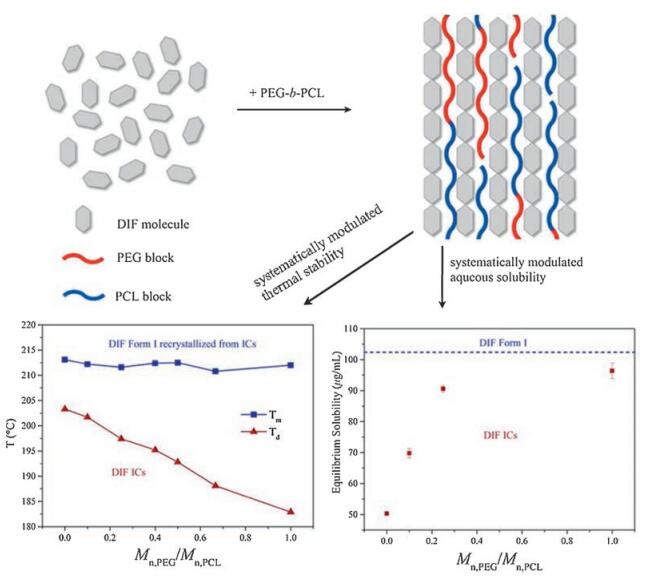
As reported in our previous publication [16], DIF/PCL IC showed a phase transition to DIF Form I during heating, which was associated with the dissociation of the guest polymer and the simultaneous recrystallization of DIF. Similar phenomena were observed for DIF/PEG-b-PCL ICs in this study. As shown in Fig. 6, DIF/PEG-b-PCL ICs all exhibited an endothermal event before the final fusion in the DSC heating scan, which was attributed to the transformation to DIF Form I as confirmed by POM observation (Fig. S7 in Supporting information). This endothermal event varied with the guest copolymers, displaying a strong dependence on the length of the PCL block. Increase of the molecular weight of the PCL block from 2 kDa to 20 kDa (decrease of Mn, PEG/Mn, PCL from 1.00 to 0.10) caused a significant increase in the dissociation temperature from 182.9 ℃ to 201.7 ℃, indicating the enhanced thermal stability of the IC crystals with the increased PCL block length. When the PCL homopolymer was used as the guest polymer, the resulting IC crystals exhibited the highest dissociation temperature in this study. The dissociation temperatures of these IC crystals as well as the melting points of the recrystallized DIF Form I crystals are listed in Table 3. It should be noted that, the recrystallized DIF Form I crystals all showed slightly lower melting points compared with that of the raw material (~214 ℃, Fig. S2), which can be ascribed to the melting point depression commonly observed in drug-polymer systems [14, 41, 42].

|
Download:
|
| Fig. 6. DSC thermograms of DIF/PEG-b-PCL ICs, measured at a heating rate of 10 ℃/ min. The endothermal events corresponding to the dissociation of the ICs are marked by the red dotted wireframe. | |
|
|
Table 3 Dissociation temperatures (Td) of the DIF ICs and melting points (Tm) of the DIF Form I crystals recrystallized from ICs. |
Since PCL and PEG used in this study have similar melting temperatures (Fig. S2), the variation of the thermal stability of DIF ICs with the block length probably reflects the different strength of the drug-polymer interactions in these IC crystals, though they possess isostructural crystal lattices. The interactions of the DIF nanochannels with the PCL block seem to be stronger than those with the PEG block. Therefore, longer PCL blocks would bring stronger drug-polymer interactions and consequently enhanced thermal stability of the IC crystals. This conclusion is consistent with the tendency for IC formation described above, where longer PCL blocks facilitated the formation of DIF ICs possibly due to the stabilization of the IC channels from the inclusion of the PCL block.
In addition to thermal stability, dissolution behavior is another important issue that needs to be carefully considered for drug solid forms. The powder dissolution profiles of DIF/PEG-b-PCL ICs with various block lengths are shown in Fig. 7. For comparison, the dissolution profiles of DIF/PCL IC and DIF Form I were also determined under the same conditions.

|
Download:
|
| Fig. 7. Powder dissolution profiles of the DIF ICs and DIF Form I at pH 4.4 under nonsink conditions (N = 3). | |
It can be seen that the DIF ICs exhibited guest-dependent dissolution profiles, which were all different from that of the pure DIF Form I crystals. For DIF/PEG-b-PCL ICs, the dissolution rate and apparent plateau solubility decreased with the increased PCL block length. The lowest dissolution rate and apparent plateau solubility in this study was obtained when using the PCL homopolymer as the guest. No solid phase transformation was observed for DIF/ PEG-b-PCL ICs even after 4 days in the dissolution media (Figs. S8 and S9 in Supporting information), suggesting that they are likely the thermodynamically stable forms when left alone in the dissolution media, similar to DIF/PCL IC and DIF Form I under the same conditions [20].
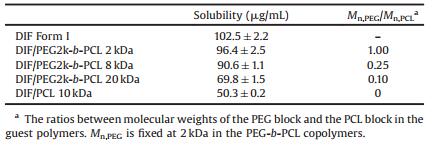
The equilibrium solubility of the DIF ICs and DIF Form I was determined by suspending the crystals in the dissolution media for 4 days, as listed in Table 4. The DIF ICs all showed lower solubility than pure DIF crystals. A significant decrease in the equilibrium solubility was observed for the DIF ICs by reducing Mn, PEG/Mn, PCL in the guest polymers. This trend in solubility of the DIF ICs can be rationalized in terms of the strength of the crystal lattice (as indicated by the thermal stability of the IC crystals) and the hydrophobicity of the crystal components (as indicated by the block lengths of the guest polymers), which are the two key factors in controlling the aqueous solubility of a crystal [5]. Therefore, DIF ICs with lower Mn, PEG/Mn, PCL values would possess stronger crystal lattice and more hydrophobic crystal components, and consequently decreased aqueous solubility.
|
|
Table 4 Equilibrium solubility of DIF Form I and the DIF ICs at pH 4.4. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
From the dissolution profiles (Fig. 7), it seems that the release of DIF from its Form I crystals is slower than those from DIF/PEG-b-PCL ICs, even though DIF Form I possesses higher aqueous solubility than these IC crystals. This phenomenon can be explained by the different morphologies of these DIF crystals (Fig. S10 in Supporting information). DIF Form I crystals displayed the characteristic monolith-like morphology, while the IC crystal particles were actually made up of smaller crystalline subparticles, which generated larger dissolution areas and consequently faster drug release, though they possessed the same gross particle size range. In addition, though PEG-b-PCL is an amphiphilic block copolymer, no micelle formation was observed during the dissolution of DIF/PEG-b-PCL ICs. Coalescence and crystallization of the hydrophobic PCL blocks occurred in the aqueous media (Fig. S11 in Supporting information), similar to that observed for DIF/PCL IC [20].
Combining all the aforementioned results, it can be concluded that the thermal stability and aqueous solubility of DIF crystals can be systematically modulated by forming isostructural ICs with PEG-b-PCL of various block lengths, as schematically summarized in Fig. 8. The decrease of Mn, PEG/Mn, PCL in the guest polymers strengthens the crystal lattice of the ICs and consequently produces higher thermal stability. The strength of the crystal lattice and the hydrophobicity of the guest polymers increase monotonically with reducing Mn, PEG/Mn, PCL, making the aqueous solubility of the IC crystals decrease in a controllable manner.

|
Download:
|
| Fig. 8. Schematic diagram showing the systematic modulation of the thermal stability and aqueous solubility of the DIF crystals by forming isostructural ICs with PEG-b-PCL of various block lengths. | |
{kind=link}
3. Conclusion
In this study, we extended the guest of drug/polymer inclusion complexes from homopolymers to block copolymers. DIF/PEG-b-PCL ICs were used as the model system to demonstrate the influence of the amphiphilic guest copolymer, PEG-b-PCL, on the formation, thermal stability and dissolution behavior of drugpolymer ICs. The tendency of IC formation was found to be correlated with the PCL block length, with longer PCL blocks facilitating the formation of the IC crystals. The resulting DIF/PEGb-PCL ICs showed an isostructural nature with our previously studied DIF/PCL IC, associated with the simultaneous inclusion of both the PEG and PCL block. Slight expansion of the channel structure was also observed compared with DIF/PCL IC, probably due to the incorporation of the PEG block. Both the blocks residing in the isolated DIF nanochannels adopted extended conformations similar to those in their bulk crystalline materials.
The effect of the block length on the thermal stability and dissolution behavior of DIF ICs was also investigated. The reduction of Mn, PEG/Mn, PCL in the guest polymers strengthened the crystal lattice of the ICs and consequently improved their thermal stability. The DIF ICs exhibited gradually reduced aqueous solubility with decreasing Mn, PEG/Mn, PCL, as a result of the simultaneous increase in the strength of the crystal lattice and the hydrophobicity of the crystal components. Therefore, the formation of DIF/PEG-b-PCL ICs extends the possible guest candidates for drug-polymer ICs from homopolymers to copolymers, and represents a new strategy to tune the desired pharmaceutical profiles of drugs in a predictable and controllable manner.
4. Experimental 4.1. MaterialsDiflunisal (DIF Form I, 98% purity), poly(ε-caprolactone) (PCL, Mn 10 kDa) and poly(ethylene glycol) (PEG, Mn 2 kDa) were obtained from Sigma-Aldrich. Block copolymer PEG-b-PCL, with the fixed PEG block length (Mn 2 kDa) and various PCL block lengths (Mn 0.5 kDa, 1 kDa, 2 kDa, 3 kDa, 4 kDa, 5 kDa, 8 kDa and 20 kDa), were purchased from KERUIDI Co., Ltd. (China). All the solvents used in this study were AP grade and used without further purification. The preparation of DIF/PCL IC followed the method described in our previous publication [16]. For the guest polymers containing PEG block, the feeding amounts of the guest polymers for the cocrystallization process were calculated by transforming the PEG block into PCL block with equivalent number of bonds in their main chains, i.e., each EG unit equals to 3/7 CL unit. Appropriate amounts of DIF and PEG-b-PCL were dissolved in a solvent mixture (ethyl acetate/heptane, 4/1, v/v), and stirred at 45 ℃ for 10 min to ensure complete dissolution. The solution was then allowed to evaporate slowly at room temperature for 3 days. White crystals formed, and were collected and dried under vacuum at room temperature for 24 h.
4.2. Powder X-ray diffraction (PXRD)Before diffraction measurements, the samples were gently ground with an agate mortar and pestle, and then sieved through a 200 mesh sieve. The tests were conducted at room temperature on a Rigaku D/max-2500 X-ray diffractometer with Cu Kα radiation (1.5418 Å). Continuous scans were performed at a speed of 2°/min and 0.02° per step in the range 2θ = 2-45°.
4.3. Solution NMRSolution 1H NMR was used to determine the chemical composition of DIF/PEG-b-PCL ICs. Appropriate amounts of the IC crystals were dissolved in deuterated acetone and 1H NMR spectra were collected using a JOEL JNM-ECA300 nuclear magnetic resonance spectrometer. The molar ratios of DIF to the monomer units in the IC crystals were calculated using the integral area of the characteristic peaks corresponding to the different protons of DIF and the guest copolymers.
4.4. Solid-state NMR (ssNMR)The 1H and 13C ssNMR experiments were performed on a Bruker AVANCE Ⅲ 600 WB spectrometer operating at 600.44 MHz and 150.92 MHz for 1H and 13C, respectively. A 4 mm double resonance MAS probe was used for the 13C experiments. In the 2D 1H-13C HETCOR experiment, the FSLG decoupling was applied during t1 (the 1H dimension) and the proton RF field was set to 100 kHz. The contact time in the 1H-13C CP process was 1 ms in this experiment. The MAS speed was 10 kHz.
4.5. Differential scanning calorimetry (DSC)DSC analysis was conducted on a Shimadzu DSC-60 differential scanning calorimeter. Temperature calibration was carried out using an indium metal standard before test. 3-5 mg powder samples were sealed in aluminum pans and analyzed from -15 ℃ to 240 ℃ at a heating rate of 10 ℃/min using a similar empty pan as the reference. An inert atmosphere was maintained in the calorimeter by purging nitrogen gas at a flow rate of 50 mL/min.
4.6. Thermogravimetric analysis (TGA)TGA tests were conducted on a Shimadzu DTG-60 apparatus. Powder samples (7-10 mg) were placed in aluminum pans, and heated over the temperature range of 25-450 ℃, at a heating rate of 10 ℃/min. Nitrogen gas of a flow rate of 50 mL/min was purged into the sample chamber to create an inert atmosphere during the experiment.
4.7. Scanning electron microscopy (SEM)The SEM images of the as prepared and partially dissolved (4 h) DIF ICs and DIF Form I particles were obtained using a fieldemission electron microscope (SU8000, HITACHI), with an accelerating voltage of 10 kV.
4.8. Polarized optical microscopy (POM)Polarized optical microscope (BX41, Olympus) equipped with a digital camera (Moticam Pro 282A, Motic) and a hot stage (LTS420, Linkam), was used to observe the thermal behavior of DIF ICs at a heating rate of 10 ℃/min. The changes in particle morphology of DIF ICs in the dissolution process were also observed in-situ under POM. A small amount of IC particles was placed between two coverslips, which were immersed in pH 4.4 buffer in a Petri dish. The morphology of a single particle was recorded at regular time intervals.
4.9. Solubility and dissolution profilesSince DIF/PCL IC is a metastable form at the high pH value [20], the dissolution experiments of DIF ICs in this study were conducted at pH 4.4 to avoid any possible solid phase transformation during the dissolution process. For comparison, the dissolution profile of DIF Form I, which is the thermodynamically stable polymorph of DIF under ambient conditions [27], was also tested under the same conditions. The dissolution behavior of DIF crystals was studied in the phosphate-citrate buffer of pH 4.4. The buffer was prepared by mixing aqueous disodium hydrogen phosphate solution (0.2 mol/L) and aqueous citric acid solution (0.1 mol/L), with a volume ratio of 8.82:11.18. All the solid samples were gently ground and sieved, and the particles in the size range of 150-180 mm were collected and used in the dissolution experiments. The dissolution profiles were obtained by monitoring DIF concentration using UV-vis spectrophotometer (Agilent-8453, Agilent Technologies) at room temperature. The absorbance-concentration curve of DIF in the buffer was established using the absorption peak at wavelength 252 nm.
The dissolution tests were conducted under nonsink conditions (i.e., where excess solids would remain at the final equilibrium state). Two hundred milligrams of DIF Form I or appropriate amount of DIF ICs (containing 200 mg of DIF equivalent) was added to 500 mL of the buffer. The added solid amount represented 3.9 times the solubility of DIF Form I in the dissolution media. The resulting suspension was stirred at a constant speed of 400 rpm (MS-PA, Scilogex). Three milliliter of the suspension was removed from the dissolution media and replaced by 3 mL of the buffer at regular time intervals. The removed suspension was filtered (cellulose acetate-nitrocellulose membrane, 0.22 mm), and the first 1 mL of the filtrate was discarded. The UV-vis absorption spectra of the remaining filtrate were collected using a quartz cuvette with an optical length of 1 cm. The absorption peak at 252 nm was used to determine the DIF concentration.
The equilibrium solubilities of DIF Form I and DIF ICs in the dissolution media were determined by the following procedure. Excess amounts of DIF Form I or DIF ICs were suspended in the phosphate-citrate buffer and stirred for 4 days before filtration. The first 2 mL of the filtrate was discarded and the remaining filtrate was diluted proper times to determine the DIF concentration by UV-vis absorption. All the above experiments were repeated for 3 times, and the residual solids were collected to check for the possible crystal transformation.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21434008, 21374054) and National Basic Research Program of China (973 Program, No. 2014CB932202).
| [1] | C.R. Gardner, C.T. Walsh, O. Almarsson. Drugs as materials:valuing physical form in drug discovery. Nat. Rev. Drug Discovery 3(2004)926–934. DOI:10.1038/nrd1550 |
| [2] | Y. Kawabata, K. Wada, M. Nakatani, S. Yamada, S. Onoue. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system:basic approaches and practical applications. Int. J. Pharm. 420(2011)1–10. DOI:10.1016/j.ijpharm.2011.08.032 |
| [3] | C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46(2001)3–26. DOI:10.1016/S0169-409X(00)00129-0 |
| [4] | C.A. Lipinski. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44(2000)235–249. DOI:10.1016/S1056-8719(00)00107-6 |
| [5] | H.D. Williams, N.L. Trevaskis, S.A. Charman, et al., Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 65(2013)315–499. DOI:10.1124/pr.112.005660 |
| [6] | M. Pudipeddi, A.T.M. Serajuddin. Trends in solubility of polymorphs. J. Pharm. Sci. 94(2005)929–939. DOI:10.1002/jps.20302 |
| [7] | G.C. Viscomi, M. Campana, M. Barbanti, et al., Crystal forms of rifaximin and their effect on pharmaceutical properties. CrystEngComm 10(2008)1074–1081. DOI:10.1039/b717887e |
| [8] | N.J. Babu, A. Nangia. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 11(2011)2662–2679. DOI:10.1021/cg200492w |
| [9] | N. Schultheiss, A. Newman. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth Des. 9(2009)2950–2967. DOI:10.1021/cg900129f |
| [10] | G. Bolla, A. Nangia. Pharmaceutical cocrystals:walking the talk. Chem. Commun. 52(2016)8342–8360. DOI:10.1039/C6CC02943D |
| [11] | N.K. Duggirala, M.L. Perry, O. Almarsson, M.J. Zaworotko. Pharmaceutical cocrystals:along the path to improved medicines. Chem. Commun. 52(2016)640–655. DOI:10.1039/C5CC08216A |
| [12] | A.T.M. Serajuddin. Salt formation to improve drug solubility. Adv. Drug Delivery Rev. 59(2007)603–616. DOI:10.1016/j.addr.2007.05.010 |
| [13] | A. Avdeef. Solubility of sparingly-soluble ionizable drugs. Adv. Drug Delivery Rev. 59(2007)568–590. DOI:10.1016/j.addr.2007.05.008 |
| [14] | Y. Huang, W.G. Dai. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharmacol. Sin. B 4(2014)18–25. DOI:10.1016/j.apsb.2013.11.001 |
| [15] | S. Janssens, G. Van den Mooter. Review:physical chemistry of solid dispersions. J. Pharm. Pharmacol. 61(2009)1571–1586. DOI:10.1211/jpp.61.12.0001 |
| [16] | Z. Zhong, C. Guo, X. Yang, et al., Drug molecule diflunisal forms crystalline inclusion complexes with multiple types of linear polymers. Cryst. Growth Des. 16(2016)1181–1186. DOI:10.1021/acs.cgd.6b00010 |
| [17] | Z. Zhong, C. Guo, L. Chen, J. Xu, Y. Huang. Co-crystal formation between poly (ethylene glycol) and a small molecular drug griseofulvin. Chem. Commun. 50(2014)6375–6378. DOI:10.1039/C4CC00159A |
| [18] | C.C. Sun. Novel Co-crystals between polyethylene glycols and 5-phenylpyrazolyl-1-benzene-sulfonamides. PCT Pat. Appl. (2006)2006/024930 A1. |
| [19] | X. Yang, Z. Zhong, Y. Huang. The effect of PEG molecular weights on the thermal stability and dissolution behaviors of griseofulvin-PEG crystalline inclusion complexes. Int. J. Pharm. 508(2016)51–60. DOI:10.1016/j.ijpharm.2016.05.014 |
| [20] | Z. Zhong, X. Yang, B. Guo, J. Xu, Y. Huang. Dissolution behavior of the crystalline inclusion complex formed by the drug diflunisal and poly (ε-caprolactone). Cryst. Growth Des. 17(2017)355–362. DOI:10.1021/acs.cgd.6b01578 |
| [21] | F.E. Porbeni, I.D. Shin, X. Shuai, et al., Morphology and dynamics of the poly (ε-caprolactone)-b-poly(L-lactide)diblock copolymer and its inclusion compound with α-cyclodextrin:A solid-state 13C NMR study. J. Polym. Sci. Part B:Polym. Phys. 43(2005)2086–2096. DOI:10.1002/(ISSN)1099-0488 |
| [22] | J. Lu, I.D. Shin, S. Nojima, A.E. Tonelli. Formation and characterization of the inclusion compounds between poly(ε-caprolactone)-poly(ethylene oxide)-poly(ε-caprolactone)triblock copolymer and α-and γ-cyclodextrin. Polymer 41(2000)5871–5883. DOI:10.1016/S0032-3861(99)00773-9 |
| [23] | J. Li, X. Ni, Z. Zhou, K.W. Leong. Preparation and characterization of polypseudorotaxanes based on block-selected inclusion complexation between poly (propylene oxide)-poly(ethylene oxide)-poly (propylene oxide) triblock copolymers and α-cyclodextrin. J. Am. Chem. Soc. 125(2003)1788–1795. DOI:10.1021/ja026623p |
| [24] | X. Li, J. Li, K.W. Leong. Role of intermolecular interaction between hydrophobic blocks in block-selected inclusion complexation of amphiphilic poly (ethylene oxide)-poly. Polymer 45(2004)6845–6851. DOI:10.1016/j.polymer.2004.07.038 |
| [25] | S. Bracco, A. Comotti, L. Ferretti, P. Sozzani. Supramolecular aggregation of block copolymers in the solid state as assisted by the selective formation of inclusion crystals. J. Am. Chem. Soc. 133(2011)8982–8994. DOI:10.1021/ja201551n |
| [26] | N. Vasanthan, I.D. Shin, L. Huang, S. Nojima, A.E. Tonelli. Formation, characterization, and segmental mobilities of block copolymers in their urea inclusion compound crystals. Macromolecules 30(1997)3014–3025. DOI:10.1021/ma970213h |
| [27] | M.C. Martínez-Oha'rriz, C. Martín, M.M. Goñi, et al., Polymorphism of diflunisal:Isolation and solid-state characteristics of a new crystal form. J. Pharm. Sci. 83(1994)174–177. DOI:10.1002/jps.2600830212 |
| [28] | W.I. Cross, N. Blagden, R.J. Davey. A whole output strategy for polymorph screening:combining crystal structure prediction, graph set analysis, and targeted crystallization experiments in the case of diflunisal. Cryst. Growth Des. 3(2003)151–158. DOI:10.1021/cg025589n |
| [29] | L.K. Hansen, G.L. Perlovich, A. Bauer-Brandl. Diflunisal-hexane (4/1). Acta Crystallogr, Sect. E:Struct. Rep. Online 57(2001)o604–o606. DOI:10.1107/S1600536801008558 |
| [30] | L.K. Hansen, G.L. Perlovich, A. Bauer-Brandl. The 1:1 hydrate of diflunisal. Acta Crystallogr, Sect. E:Struct. Rep. Online 57(2001)o477–o479. DOI:10.1107/S1600536801006973 |
| [31] | P. Sozzani, A. Comotti, S. Bracco, R. Simonutti. Cooperation of multiple CH…π interactions to stabilize polymers in aromatic nanochannels as indicated by 2D solid state NMR. Chem. Commun. 7(2004)768–769. |
| [32] | S. Bracco, A. Comotti, P. Valsesia, M. Beretta, P. Sozzani. Self-assembly of 1, 4-cis-polybutadiene and an aromatic host to fabricate nanostructured crystals by CH…π interactions. CrystEngComm 12(2010)2318–2321. DOI:10.1039/c002931a |
| [33] | X. Yan, B. Peng, B. Hu, Q. Chen. PEO-urea-LiTFSI ternary complex as solid polymer electrolytes. Polymer 99(2016)44–48. DOI:10.1016/j.polymer.2016.06.056 |
| [34] | L. Kobr, K. Zhao, Y. Shen, J. Michl, et al., Inclusion compound based approach to arrays of artificial dipolar molecular rotors. A surface inclusion. J. Am. Chem. Soc. 134(2012)10122–10131. DOI:10.1021/ja302173y |
| [35] | D.L. Vanderhart. Influence of molecular packing on solid-state 13C chemical shifts:the n-alkane. J. Magn. Reson. 44(1981)117–125. |
| [36] | E. Pe'rez, D.L. Vanderhart. Solid-state 13C nuclear magnetic resonance investigation of poly(oxetanes):effect of chain conformation. Polymer 28(1987)733–738. DOI:10.1016/0032-3861(87)90221-7 |
| [37] | A.E. Tonelli. Are the steric effects on the 13C-NMR chemical shifts of hydrocarbon polymers really long range. Macromolecules 12(1979)255–256. DOI:10.1021/ma60068a017 |
| [38] | H. Bittiger, R.H. Marchessault, W.D. Niegisch. Crystal structure of poly-ε-caprolactone, Acta Crystallogr. Sect. B:Struct. Crystallogr. Cryst. Chem. 26(1970)1923–1927. DOI:10.1107/S0567740870005198 |
| [39] | H. Tadokoro, Y. Chatani, T. Yoshihara, S. Tahara, S. Murahashi. Structural studies on polyethers, [-(CH2)m-O-]n. Ⅱ. Molecular structure of polyethylene oxide. Makromol. Chem. 73(1964)109–127. |
| [40] | Y. Takahash, I. Sumita, H. Tadokoro. Structural studies of polyethers. Ⅸ. Planar zigzag modification of poly(ethylene oxide). J. Polym. Sci., Part B:Polym. Phys. 11(1973)2113–2122. DOI:10.1002/pol.1973.180111103 |
| [41] | J. Tao, Y. Sun, G.G.Z. Zhang, L. Yu. Solubility of small-molecule crystals in polymers:D-mannitol in PVP, indomethacin in PVP/VA, and nifedipine in PVP/VA. Pharm. Res. 26(2009)855–864. DOI:10.1007/s11095-008-9784-z |
| [42] | D. Lin, Y. Huang. A thermal analysis method to predict the complete phase diagram of drug-polymer solid dispersions. Int. J. Pharm. 399(2010)109–115. DOI:10.1016/j.ijpharm.2010.08.013 |