 2017, Vol. 28
2017, Vol. 28 



b Key Laboratory of Pesticide Chemistry and Application, Ministry of Agriculture, Department of Applied Chemistry, College of Science, China Agricultural University, Beijing 100193, China;
c Central Research Institute of China Chemical Science & Technology, Beijing 100083, China
Fungal infections are a worldwide threat to human health and food security [1, 2]. However, drug resistance risks are on the rise. Thus, there is a substantial and constant need for novel antifungal agents.
Chitin, a polymer of β-1, 4 conjugate N-acetylglucosamine, is an essential structural polysaccharide found in the cell wall and septa of all pathogenic fungi but not in plants and vertebrates [3]. Thus, chitin synthase (CHS), the key enzyme responsible for catalyzing chitin biosynthesis, is an attractive target for new fungicides [4, 5].
CHS (EC 2.4.1.16) belongs to the glycosyltransferase family 2 (GT2) and is responsible for catalyzing the transfer of a N-acetyl-D-glucosamine (GlcNAc) residue from UDP-N-acetyl-D-glucosamine (UDP-GlcNAc) to nascent chitin chain [6]. This reaction is presumed to include the formation of a donor-acceptor intermediate (Scheme 1) [7-9]. A recent crystallographic study of the GT2 bacterial cellulose synthase (BcsA) resolved both the donor UDPsugar binding site and the acceptor nascent cellulose chain binding site [10]. Base on this structure of BcsA, the structure of a chitin synthase (NodC) from the bacteria Rhizobium leguminosarumi was modelled. The structure-function analysis of NodC indicates that CHS has a similar catalytic pocket as BcsA [11]. Thus, this conserved catalytic pocket structure can be used for designing new CHS inhibitors.

|
Download:
|
| Scheme 1. Presumed catalytic mechanism of CHS. | |
Previous inhibitors against fungal CHS have been isolated from natural sources or obtained from large chemical libraries [12-16]. Polyoxins and nikkomycins are potent inhibitors isolated from Streptomyces genus, which show high structural similarity with the donor UDP-GlcNAc. These competitive inhibitors were derived from the scaffold of a peptide linkage between an UDP analogue and a sugar mimicry moiety [17]. Structural optimization of these nucleoside-peptide compounds were performed in all the three parts: uridine moiety, peptide linkage, and sugar mimicry moiety. The isopropylidene modified uridine derivatives were synthesized and showed similar activity with nikkomycin Z [18]. Interestingly, Jean-Bernard Behr et al. used a series of diphosphate bioisosteres to mimic the presumed diphosphate-M2+ intermediate formed in the active site, but only weak CHS inhibition activity was obtained [19]. However, optimization of the sugar mimicry moiety can improve the inhibitory activity. When a phenanthrene group was added to the terminal amino acid of nikkomycin Z, CHS inhibitory activity was slightly increased [20]. Furthermore, using an alky chain to replace the sugar mimicry moiety of polyoxin L, a wellknown CHS inhibitor, to increase its hydrophobicity enhanced its antifungal activity [21]. Moreover, an additional hydrophobic group added either to the peptide linkage or the sugar mimicry moiety increased binding affinity [22-25]. Taken together, modification of the donor UDP-sugar is a feasible strategy for developing novel CHS inhibitors.
In this work, we designed and synthesized a series of compounds, by modifying all three parts of the donor substrate UDP-GlcNAc: sugar, di-phosphate, and uridine moieties. Based on the modelled CHS structure, novel UDP-sugar analogues with an extra hydrophobic chain on the linkage between the sugar mimicry and the N-acylhydrazone moiety were designed. These newlydesigned molecules exhibited both high inhibitory activities against CHS and antifungal activities against the plant-parasitic fungi, F. graminearum, B. cinerea, and C. lagenarium. Thus, we provide a promising scaffold for developing novel fungicides.
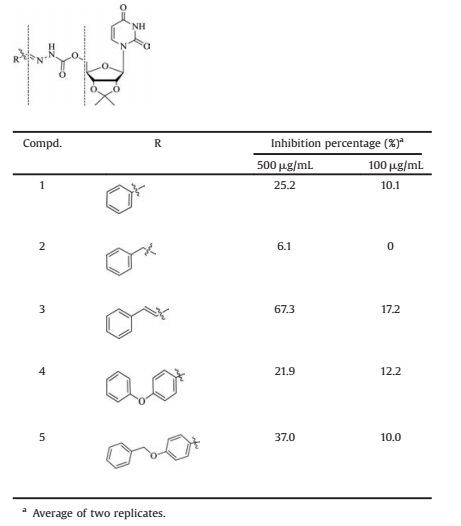
2. Results and discussion 2.1. Modification of the UDP-GlcNAc scaffoldTo develop a novel scaffold, we designed, synthesized and evaluated a series of substrate donor analogs, which were modified in both their UDP and sugar moieties. The UDP moiety is composed of one di-phosphate and one uridine. Based on the crystal structure of BcsA (pdb code: 4P00), the di-phosphate group of UDP likely binds the magnesian ion, necessary for the catalytic process. In light of this, an N-acylhydrazone group, which has the ability to bind to metals, was designed to replace the di-phosphate moiety [26]. For the uridine moiety, we introduced an isopropylidene to bridge the 2-and 3-hydroxyl groups of ribose in accordance to a CHS inhibitor design by Chaudhary et al. [18]. To optimize the sugar moiety, a benzene was chosen to mimic the GlcNAc moiety that might have stacking interaction with the enzyme. Assembling and synthesizing all of these optimized moieties led to the compound 1. Inhibitory activity assays indicated that compound 1 exhibited a weak activity of 25.2% against CHS at 500 μg/mL. To enhance the interaction between the benzene moiety and the enzyme, further optimization on the linker between the benzene and N-acylhydrazone moieties were performed (Table 1). This strategy was successful and the most potent compound, compound 3 (67.3%), exhibited a 2.7-fold inhibitory activity improvement at 500 μg/mL. The broad range of activities for compounds 1-5 further supported the importance of the linker between the benzene and N-acylhydrazone moieties for inhibitory activity. Thus, compound 3, containing an ethylene residue linker, was selected as a starting point for further modifications.
|
|
Table 1 Inhibitory activities of compounds 1–5 toward CHS. |
2.2. Design of the novel branched scaffold
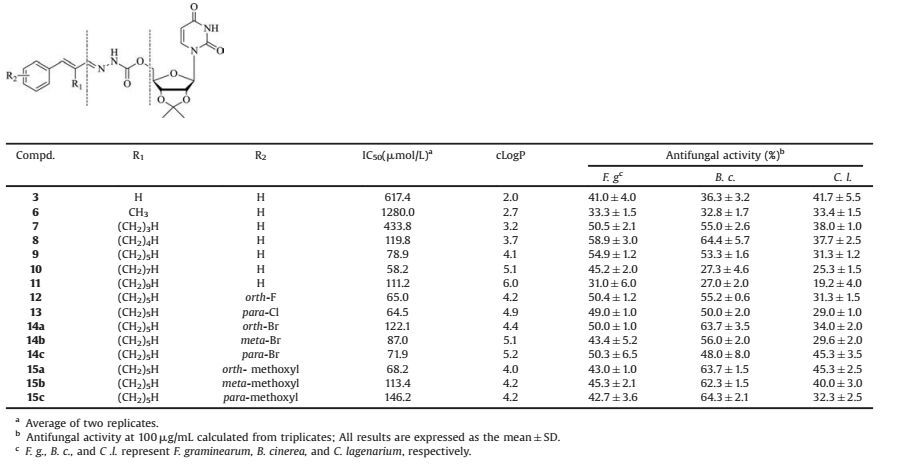
As indicated in the modelled structure of CHS, the acceptor binding site is in close proximity to the donor sugar binding site of CHS [11]. Thus, the addition of groups to the sugar mimicry moiety of the substrate donor analogs of CHS might interact with the acceptor binding site and improve the inhibitory activity. The acceptor binding site is a narrow hydrophobic channel in the homo-structure of CHS (BcsA), thus a flexible slender hydrophobic moiety would be better suited for binding this site. Based on this inference, alkyl chains were introduced to the linker between the benzene and the N-acylhydrazone moieties of compound 3 and a series of branched substrate mimicries were synthesized (compounds 6-11). These branched compounds all exhibited inhibitory activities toward CHS, with the longer alkyl chains having higher inhibitory activity, except for compound 1 (Table 2). Compound 10, with an IC50 value of 58.2 μmol/L, was the most effective CHS inhibitor. However, the antifungal activities of compound 10, against all three tested plant-parasitic fungi, were significantly lower than compound 9, which exhibited a similar CHS inhibitory activity (IC50 = 78.9 μmol/L) with compound 10. Thus, compound 9 was chosen for further optimization.
|
|
Table 2 IC50 values, cLogP values, and antifungal activities of compounds 3 and 6–15. |
{kind=link}
Based on the structure of compound 9, the benzene moiety was further optimized by introducing substitution groups on the benzene moiety (Table 2). Compounds 12 and 13 were the most potent with IC50 values of 65.0 μmol/L and 64.5 μmol/L, respectively. Compared with these two inhibitors, the inhibitory activity of either bromine or methoxyl substitution decreased, suggesting that these oversize groups might introduce steric effects, which influence the benzene moiety binding (compounds 14-15). Thus, a smaller substitution moiety on the benzene moiety benefits inhibitory activity. Interestingly, the substitution with a parabromine (compound 14c) or an orth-methoxyl (compound 15a) group on the benzene moiety of compound 9 slightly enhanced its inhibitory activity. This suggests that finding a suitable substitution position for these oversize groups may decease this steric effect, and that these improvements might mainly be the result of orientation effects. Furthermore, halogen substitution on the orth-or para-position, except for compound 14a, enhanced the binding affinity (compounds 12-14). Meanwhile, substitution with bromine atom on the meta-position was adverse to inhibitory activity (compound 14b).
2.3. Antifungal activity of CHS inhibitors 6-15As chitin synthases are essential for fungi survival, we deduced that these chitin synthases inhibitors can inhibit fungi growth. All of these branched CHS inhibitors exhibited antifungal activities against growth of the destructive fungi, F. graminearum, B. cinerea, and C. lagenarium. Compound 8, with 58.9% ± 3.0% growth inhibition of F. graminearum, had similar effectiveness as the commercial fungicide, polyoxin B (57.4% ±2.5%) at 100 μg/mL. Some other compounds (compounds 7-9, 12, 14a, and 14c) also inhibited the growth of F. graminearum more than 50%. Furthermore, B. cinerea was also sensitive to these synthetic compounds with most compounds (compounds 7, 8, 12, 13, 14a, 14b, and 15a-c) exhibiting even stronger growth inhibition against B. cinerea than F. graminearum. However, this series of compounds was less effective against C. lagenarium.
Interestingly, high hydrophobicity of compounds 9-11, with a cLogP value more than 3.7, adversely affected antifungal activity. This inference was also observed in the compounds with a bromine substitution. Compound 14a (orth-Br, IC50 = 122.1 μmol/L, cLogP = 4.4) exhibited a higher antifungal activity than the more hydrophobic compound 14b (meta-Br, IC50 = 87.0 μmol/L, cLogP = 5.1) even though it possessed a lower inhibitory activity towards CHS. This could potentially be due to solubility of the compounds or inability to penetrate the cell wall. Moreover, 14c (para-Br, IC50 = 71.9 μmol/L, cLogP = 5.2) had a similar antifungal activity as 14a, despite having nearly twice the inhibitory activity, further demonstrating the importance of hydrophobicity.
3. ConclusionsThe donor substrate UDP-GlcNAc of CHS is a good starting point for CHS inhibitors development. Based on the structure of UDPGlcNAc, as well as the modelled CHS structure, we developed a novel scaffold of CHS inhibitors. All inhibitors bearing this novel scaffold exhibited antifungal activities against growth of destructive fungus F. graminearum, B. cinerea and C. lagenarium. Altogether, the scaffold developed in this work shows promise for the development of novel fungicides.
4. Materials and methods 4.1. Instruments and reagentsMelting points were measured on a Cole-Parmer melting point apparatus, and the thermometer was uncorrected. 1H NMR spectra were obtained using a Bruker Advance DPX (300 MHz) (Switzerland) with tetramethylsilane and hexadeuterodimethyl sulfoxideas as the internal standard and solvent, respectively. Elemental analysis was performed on a Vario EL Ⅲ Flash EA 1112 elemental analyzer by the Analytical Center of the Institute of Chemistry, Chinese Academy of Science. Infrared spectra were recorded using a PerkinElmer Spectrum 100 FT-IR. Chemical reagents used for CHS activity assays were purchased from Sigma Co., and all other solvents and reagents were purchased from the Beijing Chemical Reagents Co., China.
4.2. Chemical synthesisGeneral procedure for the synthesis of target compound 1. Compound 5'-O-carbazoyl-2', 3'-O-isopropylidene (compound Ⅳ) was prepared in a three steps synthesis from uridine (compound Ⅰ) according to the published procedure [27] (Scheme 2). Compound Ⅳ (2.50 mmol), phenyl aldehyde (2.75 mmol), and 30 mL acetic acid were added into 50 mL ethanol, and the reaction mixture was stirred at room temperature for 0.5 h. Crude products were purified by flash chromatography (Vpetroleumether/Vethylacetate = 1:1) to obtain the compound 1.

|
Download:
|
| Scheme 2. General synthetic procedure for target compounds 1–5. | |
{kind=link}
Compounds 2-5 were obtained according to the synthesis procedure used to synthesize compounds 1 and their structures were confirmed by IR, 1H NMR, and elemental analysis. The data of their yields, melting points, and elemental analyses are listed in Table S1 (Supporting information) and the 1H NMR and IR data are listed in Table S2 (Supporting information).
General procedure for the synthesis of compounds α-alkylaromatic-acroleins (Ⅶ). 20 mmol Aromatic aldehyde, 20 mmol Cs2CO3, and 10 mmol benzyltriethylammonium chloride were added to a sealed 50 mL kolle flask. Then 15 mL DCM was added under N2 (using Schlenk line technology). The reaction mixture was stirred at 25 ℃ and 30 mmol alkyl aldehyde was added dropwise into the reaction mixture over 3-5 h. The reaction progression was monitored using GC, and stirred at 25 ℃ until completion. After reaching completion the reaction mixture was diluted with 50 mL DCM, washed twice with 20 mL water and once with 10 mL brine. The organic phase was collected, dried over MgSO4, filtered, and then evaporated under vacuum. The resulting crude product was used without further purification.
General procedure for the synthesis of compounds Ⅷ. The synthesis procedure used to synthesize compounds 1 was performed to obtain compounds Ⅷ by using α-alkyl aromatic acroleins (Scheme 3). The structures of compounds 6-15 were confirmed by IR, 1H NMR, and elemental analysis. The yields, melting points, and elemental analyses are listed in Table S1 (Supporting information) and the 1H NMR and IR data are listed in Table S2 (Supporting information).

|
Download:
|
| Scheme 3. General synthetic procedure for target compounds 6–15. | |
{kind=link}
Physical and elemental data of compounds Ⅴ and Ⅷ are listed in Table S1. 1H NMR and IR data of compounds Ⅴ and Ⅷ Are listed in Table S2.
4.3. Inhibitory activity assayThe chitin synthase inhibition assay was performed using yeast (Saccharomyces cerevisiae) cell extracts according to the method described by Ke et al. [28]. Briefly, yeast cells were harvested at 1500 g for 10 min after growing overnight in yeast extract peptone dextrose (YPD) media at 30 ℃. Then the harvested cells were disrupted by grinding with glass beads in liquid nitrogen. After digestion by 0.08 mg/mL trypsin at 30 ℃ for 30 min, 0.12 mg/mL soybean trypsin inhibitor was added to the cell extracts. All test samples were dissolved in DMSO. Chitin synthase assays were performed in pretreated WGA-coated (wheat germ agglutinin coated) 96-well microtiter plates using 48 μL trypsin pretreated cell extracts, 2 μL test sample or DMSO as control, and 50 μL premixed solution (80 mmol/L GlcNAc, 8 mmol/L UDP-GlcNAc, and 3.2 mmol/L CoCl2 in 50 mmol/L Tris-HCI buffer, pH 7.5). Plates were incubated on a shaker at 30-℃ for 3 h. Then plates were immediately washed 5 times with double distilled water, followed by the addition of 100 μL WGA-HRP (horseradish peroxidase conjugated wheat germ agglutinin) solution (1 μg/mL WGA-HRP and 20 mg/mL BSA in 50 mmol/L Tris-HCl buffer, pH 7.5). After incubation for 30 min at 30 ℃, plates were washed again for 5 times with double distilled water. After adding 100 mL fresh tetramethylbenzidine reaction reagent (0.8 mmol/L tetramethylbenzidine and 1.8% H2O2 in 100 mmol/L sodium acetate buffer, pH 3.7), the optical densities (OD) at 600 nm were detected every 2 min for 40 min. Then the reaction rates were calculated based on these OD600 values. The IC50 values were calculated by using reaction rates at seven different inhibitor concentrations with 5% DMSO. Each value was performed in duplicates.
4.4. Calculation of LogP valuesThe LogP values were calculated using CISOC-LOGP (version 1.0, developed by the Shanghai Institute of Organic Chemistry, CAS).
4.5. Fungicidal activity assayAntifungal activity was screened against three main plantparasitic fungi F. graminearum, B. cinerea and C. lagenarium according to the method described by Miao et al. [29]. All test compounds were dissolved in DMSO. A mycelial plug (0.5 cm in diameter) taken from the edge of a 4-day-old colony of strains was grown at 25 ℃ on the potato dextrose agar (PDA) plates containing either 1% DMSO with 100 μg/mL test compound or 1% DMSO for a negative control. For each plate, the colony radial growth diameter, from the edge of the plug to the edge of the colony, was measured in two perpendicular directions and averaged. Each test repeated four times. The antifungal activity was calculated as follow: Antifungal activity = (control growth diameter -test growth diameter)/control growth diameter.
AcknowledgementsWe thank Thomas Malott (Dalian University of Technology) for his contribution in the language editing of the manuscript. This work was supported by the Program for National Natural Science Funds for Distinguished Young Scholar (No. 31425021); the National Natural Science Foundation of China (No. 21472236); the Natural Science Foundation of Liaoning Province (No. 2015020782); and the Fundamental Research Funds for the Central Universities (No. DUT16TD22).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.03.030.
| [1] | M.C. Fisher, D.A. Henk, C.J. Briggs, et al., Emerging fungal threats to animal, plant and ecosystem health. Nature 484(2012)186–194. DOI:10.1038/nature10947 |
| [2] | S. Zhu, W. Wang, K. Fang, et al., Design, synthesis and antifungal activity of carbazole derivatives. Chin. Chem. Lett. 25(2014)229–233. DOI:10.1016/j.cclet.2013.10.022 |
| [3] | J.C. Kapteyn, L.L. Hoyer, J.E. Hecht, et al., The cell wall architecture of Candida albicans wild-type cells and cell wall-defective mutants. Mol. Microbiol. 35(2000)601–611. |
| [4] | H. Merzendorfer. Chitin synthesis inhibitors:old molecules and new developments. Insect Sci. 20(2013)121–138. DOI:10.1111/j.1744-7917.2012.01535.x |
| [5] | M.D. Lenardon, C.A. Munro, N.A.R. Gow. Chitin synthesis and fungal pathogenesis. Curr. Opin. Microbiol. 13(2010)416–423. DOI:10.1016/j.mib.2010.05.002 |
| [6] | H. Merzendorfer. Insect chitin synthases:a review. J. Comp. Physiol. B 1(2006)1–15. |
| [7] | L.L. Lairson, B. Henrissat, G.J. Davies, et al., Glycosyltransferases:structures, functions, and mechanisms. Annu. Rev. Biochem. 77(2008)521–555. DOI:10.1146/annurev.biochem.76.061005.092322 |
| [8] | U.M. Unligil, J.M. Rini. Glycosyltransferase structure and mechanism. Curr. Opin. Struct. Biol. 10(2000)510–517. DOI:10.1016/S0959-440X(00)00124-X |
| [9] | M. Winn, R.J.M. Goss, K. Kinura, T.D.H. Bugg. Antimicrobial nucleoside antibiotics targeting cell wall assembly:recent advances in structure-function studies and nucleoside biosynthesis. Nat. Prod. Rep. 27(2010)279–304. DOI:10.1039/B816215H |
| [10] | J.L. Morgan, J.T. McNamara, M. Fischer, et al., Observing cellulose biosynthesis and membrane translocation in crystallo. Nature 531(2016)329–334. DOI:10.1038/nature16966 |
| [11] | H.C. Dorfmueller, A.T. Ferenbach, V.S. Borodkin, Aalten D.M. van. A structural and biochemical model of processive chitin synthesis. J. Biol. Chem. 289(2014)23020–23028. DOI:10.1074/jbc.M114.563353 |
| [12] | E.I. Hwang, B.M. Kwon, S.H. Lee, et al., Obovatols, new chitin synthase 2 inhibitors of Saccharomyces cerevisiae from Magnolia obovata. J. Antimicrob. Chemother. 49(2002)95–101. DOI:10.1093/jac/49.1.95 |
| [13] | C. Niu, J.B. Qu, H.X. Lou. Antifungal bis[bibenzyls] from the Chinese liverwort Marchantia polymorpha L. Chem. Biodivers. 3(2006)34–40. DOI:10.1002/(ISSN)1612-1880 |
| [14] | X.Z. Wu, A.X. Cheng, L.M. Sun, H.X. Lou. Effect of plagiochin E an antifungal macrocyclic bis(bibenzyl), on cell wall chitin synthesis in Candida albicans. Acta Pharmacol. Sin. 29(2008)1478–1485. DOI:10.1111/j.1745-7254.2008.00900.x |
| [15] | M. Sudoh, T. Yamazaki, K. Masubuchi, H. Yamada-Okabe, et al., Identification of a novel inhibitor specific to the fungal chitin synthase. J. Biol. Chem. 257(2000)32901–32905. |
| [16] | H. Magellan, M. Boccara, T. Drujon, et al., Discovery of two new inhibitors of botrytis cinerea chitin synthase by a chemical library screening. Bioorg. Med. Chem. 21(2013)4997–5003. DOI:10.1016/j.bmc.2013.06.058 |
| [17] | P.M. Chaudhary, S.G. Tupe, M.V. Deshpande. Chitin synthase inhibitors as antifungal agents. Mini Rev. Med. Chem. 13(2013)222–236. |
| [18] | P.M. Chaudhary, S.R. Chavan, F. Shirazi, et al., Exploration of click reaction for the synthesis of modified nucleosides as chitin synthase inhibitors. Bioorg. Med. Chem. 17(2009)2433–2440. DOI:10.1016/j.bmc.2009.02.019 |
| [19] | J.B. Behr, T. Gourlain, A. Helimi, G. Guillerm. Design, synthesis and biological evaluation of hetaryl-nucleoside derivatives as inhibitors of chitin synthase. Bioorg. Med. Chem. Lett. 13(2003)1713–1716. DOI:10.1016/S0960-894X(03)00239-7 |
| [20] | K. Obi, J. Uda, K. Iwase, et al., A, Novel nikkomycin analogues:inhibitors of the fungal cell wall biosynthesis enzyme chitin synthase. Bioorg. Med. Chem. Lett. 10(2000)1451–1454. DOI:10.1016/S0960-894X(00)00256-0 |
| [21] | R.K. Khare, J.M. Becker, F.R. Naider. Synthesis and anticandidal properties of polyoxin l analogues containing α-amino fatty acids. J. Med. Chem. 31(1988)650–656. DOI:10.1021/jm00398a027 |
| [22] | A. Suda, A. Ohta, M. Sudoh, et al., Combinatorial synthesis of nikkomycin analogs on solid support. Heterocycles 55(2001)1023–1028. DOI:10.3987/COM-01-9222 |
| [23] | E. Krainer, J.M. Becker, F. Naider. Synthesis and biological evaluation of dipeptidyl and tripeptidyl polyoxin and nikkomycin analogs as anticandidal prodrugs. J. Med. Chem. 34(1991)174–180. DOI:10.1021/jm00105a026 |
| [24] | Q. Ji, D. Yang, X. Wang, et al., Design synthesis and evaluation of novel quinazoline-2, 4-dione derivatives as chitin synthase inhibitors and antifungal agents. Bioorg. Med. Chem. Lett. 22(2014)3405–3413. DOI:10.1016/j.bmc.2014.04.042 |
| [25] | Q. Ji, Z. Ge, Z. Ge, et al., Synthesis and biological evaluation of novel phosphoramidate derivatives of coumarin as chitin synthase inhibitors and antifungal agents. Eur. J. Med. Chem. 108(2016)166–176. DOI:10.1016/j.ejmech.2015.11.027 |
| [26] | M. Carcelli, D. Rogolino, A. Gatti, et al., N-acylhydrazone inhibitors of influenza virus PA endonuclease with versatile metal binding modes. Sci. Rep. 6(2016)31500. DOI:10.1038/srep31500 |
| [27] | J. Zhang, Y. Li, X. Yang, et al., Synthesis and bioactivities of nucleoside compounds containing substituted benzoyl carbamate thiourea. Chin. J. Org. Chem. 33(2013)305–311. DOI:10.6023/cjoc201210036 |
| [28] | S. Ke, F. Liu, N. Wang, et al., 1, 3, 4-Oxadiazoline derivatives as novel potential inhibitors targeting chitin biosynthesis:design, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 19(2009)332–335. DOI:10.1016/j.bmcl.2008.11.095 |
| [29] | H. Miao, J. Zhang, H. Yuan, et al., Synthesis and fungicidal activity of nucleoside compounds containing substituted benzoyl thiourea. Chin. J. Org. Chem. 32(2012)915–921. DOI:10.6023/cjoc1110111 |