 2017, Vol. 28
2017, Vol. 28 


 , Jiang Yu-Yanga,c
, Jiang Yu-Yanga,c
b The State Key Laboratory Breeding Base-Shenzhen Key Laboratory of Chemical Biology, The Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China;
c Department of Pharmacology and Pharmaceutical Sciences, School of Medicine, Tsinghua University, Beijing 100084, China
Histone deacetylase (HDAC), which removes acetyl groups from histone and non-histone proteins, plays a crucial role in regulating gene expression. Modulation of HDACs can impact the activity of a number of proteins, including tubulin, p53, and Hsp90, which are associated with a variety of cellular processes, such as cell cycle arrest, differentiation, and apoptosis [1]. Small molecule HDAC inhibitors vorinostat, romidepsin, and ponobinostat have been approved by the U.S. FDA as treatments for cutaneous T cell lymphoma (CTCL) and multiple myeloma [2-4].
Many studies have reported that interruption of signaling pathways, such as the MAPK and PI3 K/Akt cascades, can lower the threshold for HDAC inhibitors-induced cancer cell lethality [5, 6]. Vandetanib (1, Scheme 1), BMS-690514 (2, Scheme 1), neratinib (3, Scheme 1), and TAK-285 (4, Scheme 1) are 4-aminoquinazoline (or like) multitarget kinase inhibitors (Table S1 in Supporting information) which have been shown to inhibit EGFR (ErbB1), HER2 (ErbB2), VEGFR2, MAPK, and Akt activity. It is reported that treatment with HDAC inhibitor vorinostat (5, Scheme 1) synergized with vantanib in human cancer cells [6]. Recently, several studies have reported on kinase/HDAC multitarget inhibitors [7-9] and yielded satisfactory results on improving the response rates and overcoming the acquired resistance to single-targeted drugs, which proved it a feasible strategy worth to be investigated in more details. Herein, as part of our continuous efforts for developing potent multitarget anticancer agents [10-18], we report the preparation and evaluation of the antitumor activity of novel kinase/HDAC multitarget inhibitors which might offer benefits for cancer therapy.

|
Download:
|
| Scheme 1. Design strategy of novel kinase/HDAC multitarget inhibitors 6a–6h. | |
As shown in Scheme 1, by merging the core pharmacophore of HDAC inhibitors vorinostat (hydroxamic acid) and the pharmacophore of multitarget kinase inhibitors vandetanib, BMS-690514, neratinib, and TAK-285 (4-aminoquinazoline) with a 1, 2, 3-triazole as linker, compounds 6a-6h were designed. The 1, 2, 3-triazole could lead the hydroxamic acid group fit into the hydrophilic zone of kinase [19] and is a bio-isosterism of amide group in vorinostat which could help the 4-aminoquinazoline group recognize the surface outside the HDAC active pocket [20]. Compounds 6a-6h retained the core pharmacophores of kinase and HDAC inhibitors which may help maintain the inhibitory activity of these targets.
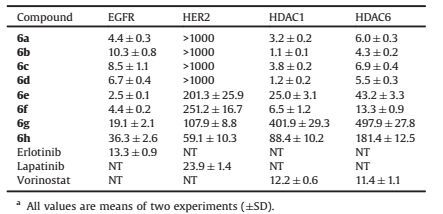
2. Results and discussionWe first tested the inhibitory activity of the synthetic hybrids against EGFR, HER2 and VEGFR-2 at the concentration of 1 μmol/L. Vandetanib, BMS-690514, neratinib, and TAK-285 are typical 4-aminoquinazoline (or like) kinase inhibitors. The structure-activity relationships (SARs) of quinazoline derivatives have been already explored for their inhibitory activities towards various protein kinase enzymes, and the key features between the kinase receptor and 4-aminoquinazoline template have been revealed as follows [19, 21]: The quinazoline moiety occupies the ATP-binding pocket in the kinase domain, while the 4-aniline ring fits into an adjacent lipophilic pocket. It is reported that different 4-anilino functional groups can alter the selectivity of quinazoline based inhibitors within different types of kinases. Substituent at C-6 of the quinazoline core extends towards the hydrophilic region, which could be utilized to improve the pharmacokinetic properties and cellular activities and is not expected to significantly affect the activity and selectivity. However, in this study, although the 4-anilino groups are retained, the selectivity profiles of compounds 6a-6h on EGFR, HER2, and VEGFR2 changed dramatically. As shown in Table 1, compounds 6a-6h maintained the outstanding potencies on EGFR, compounds 6e-6h also maintained the activity on HER2 at some extent. However, compounds 6a-6d lost the activities on HER2 and VEGFR2 totally. For instance, the reported IC50 values of vandetanib against EGFR and VEGFR2 are 500 and 40 nmol/L respectively (Table S1). After modification, compound 6a and 6b displayed more than 100-fold selectivity on EGFR and VEGFR2 (Table 1 and Table 2). These unexpected effects on selectivity profiles may result from the introduction of triazole substituted long alkyl chains of hydroxamic acid. It seems that functional groups at C-6 of the quinazoline not only have influences on the pharmacokinetic properties and cellular activities, but also affect the activity and selectivity of kinases potentially. What is interesting is that, suggested by our results, the introduction of vorinostat-like segment to quinazoline improved or maintained the inhibitions on EGFR and HDAC but reduced the inhibitions on HER2 and VEGFR2.
|
|
Table 1 Percent inhibition effects of compounds 6a–6h against VEGFR2, HER2 and EGFR at 1 μmol/La. |

Next, we tested the inhibitory activities of the target compounds against EGFR and HDAC enzymes using erlotinib and vorinostat as the positive control compounds (Table 2). Overall, the majority of the compounds exhibited outstanding potencies on the four enzymes with IC50 values in single or doubledigit nanomolar range. Given the in vitro enzymatic inhibition assay results, we ran a SAR analysis on the synthetic compounds. We first investigated the influence of chain length on the inhibitions of EGFR, HER2, and HDAC. Basically, compared to 5-carbon chain length compounds (6a, 6c, 6e, 6g), the respective 6-carbon chain length compounds (6b, 6d, 6f, 6h) possessed less than 2-fold increase or loss of inhibitory activity against EGFR and HER2. Since the long alkyl chains of hydroxamic acid fits into the hydrophilic region, the difference of one carbon chain length is not expected to have significant effects on the potency of EGFR and HER2. However, all the 6-carbon chain length compounds showed much better inhibitory activity than the respective 5-carbon chain length compounds against HDAC1 and HDAC6. Some of the 6-carbon chain length compounds even had a 5-fold enhancement of activity in inhibiting HDAC compared to the respective 5-carbon chain length compounds. which is congruent with previous reports [8]. We next evaluated the influence of the 4-aniline portion on the inhibition of EGFR, HER2, and HDAC. It is reported that the size of 4-aniline groups can influence the activity of HER2 thus alter the selectivity between EGFR and HER2 [21]. Usually, large aniline substituted quinazolines such as neratinib and TAK-285 tend to be dual-targeted inhibitors of EGFR and HER2 while small aniline substituted quinazolines such as erlotinib possess much worse potency on HER2. In this study, we found that all the compounds exhibit outstanding activities on EGFR. But in the case of HER2 inhibition, compounds 6e-6h, which possessed bigger 4-aniline groups, exhibited much better inhibitory activities than compounds 6a-6d. Different from inhibition of EGFR/HER2, 4-anilino groups are not critical for HDAC interaction. It is validated that variation of the cap group of HDAC inhibitors to a certain extent would not significantly affect their binding to HDAC. However, as the cap becomes much larger, the potency of HDAC inhibition is weakened, which may result from steric hindrance in interacting with the active pocket of HDAC. Compounds 6e-6h exhibited much worse potencies against HDAC1 and HDAC6 than compounds 6a-6d. Compound 6g had a nearly 100-fold reduction of activity in inhibiting HDAC compared to compounds 6a-6d which have the smallest substituents on the phenyl ring.
|
|
Table 2 Enzymatic inhibitory activities (IC50, nmol/L) of compounds 6a–6h against EGFR, HER2, HDAC1, and HDAC6a. |
Inhibition of HDAC could lead to upregulated acetylation of Hsp90, resulting in degradation of varies of Hsp90 client proteins such as mut-EGFR, HER2, and AKT which play important roles in cancer cell proliferation. Treatment of HDAC inhibitors vorinostat or panobinostat was reported to lead to increased acetylation of Hsp90, degradation of EGFR and HER2, and cell death in EGFR/ HER2 over-expressed cancer cell lines [22, 23]. So we next determined the anti-proliferative effects of the synthetic compounds against A549 [24, 25] cells (EGFR wild-type) and BT-474 [26] cells (HER2 overexpressed) with erlotinib and vorinostat as the positive control compounds. As shown in Table 3, compound 6b suppressed the growth of A549 cancer cells much more potently than all the other compounds including erlotinib and vorinostat, which may result from its outstanding inhibition on EGFR and HDAC. Compounds 6e-6h suppressed HER2 overexpressed BT-474 cell lines more potently than compounds 6a-6d, which is in congruent with the difference of their inhibitory potencies on HER2.
|
|
Table 3 Cellular inhibitory activities (IC50, μmol/L) of compounds 6a–6h against A549 and BT-474 cellsa. |

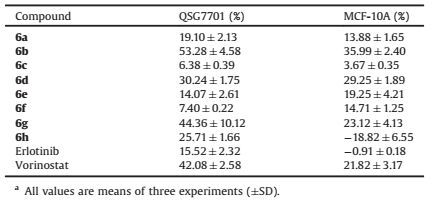
To further identify the selectivity of synthetic compounds, we evaluated the toxic activities of synthetic compounds against human normal liver cells QSG7701 [27] and human normal breast cells MCF-10A [26]. As shown in Table 4, almost all the compounds had no obvious anti-proliferative effects on these two cells at the concentration of 50 μmol/L. Compound 6b exhibited the most potent inhibitory activity on QSG7701 and MCF-10A cells with the inhibitory rate of 53.28% and 35.99% at the concentration of 50 μmol/L, which indicates that our compounds possessed quite nice safety profiles.
|
|
Table 4 Percent inhibition effects of compounds 6a–6h against QSG7701 and MCF-10A cells at 50 μmol/La. |
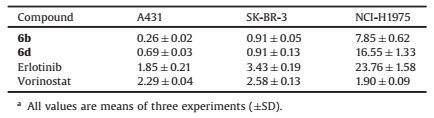
Based on the potent enzymatic activities and anti-proliferative effects of compounds 6b and 6d, we next tested their cytotoxicity on A431 (EGFR overexpressed) [28], SK-BR-3 (HER2 overexpressed) [29], and NCI-H1975 (EGFR mutated, T790 M/L858R) [25] cells. Similarly, as shown in Table 5, both compounds 6b and 6d suppressed three cell lines potently. Our results show that 6b and 6d is effective against a broad range of solid cancer cells with different levels of EGFR and HER2. Compared to 6d, 6b possessed better potencies on five cancer cell lines. Of note, 6b almost displayed comparable or greater potency in all these assays than erlotinib and vorinostat. For instance, when tested against A431 and SK-BR-3 cells, compound 6b displayed better inhibitions (IC50: 0.26 and 0.91 μmol/L on A431 and SK-BR-3 cells respectively) as compared with erlotinib (IC50: 1.85 and 3.43 μmol/L respectively) and vorinostat (IC50: 2.29 and 2.58 μmol/L respectively). compound 6b also exihibited more than 3-fold enhancement of activity than erlotinib in inhibiting erlotinib resistant NCI-H1975 cell line (with IC50 values 7.85 and 23.76 μmol/L respectively). These results suggest that 6b, with its simultaneous and synergistic inhibitions on multiple pathways, has the great potential to improve the response rates to selecive EGFR inhibitor erlotinib or selective HDAC inhibitor vorinostat.
|
|
Table 5 Cellular inhibitory activities (IC50, μmol/L) of compounds 6b and 6d against A431, SK-BR-3, and NCI-H1975 cellsa. |
{kind=link}
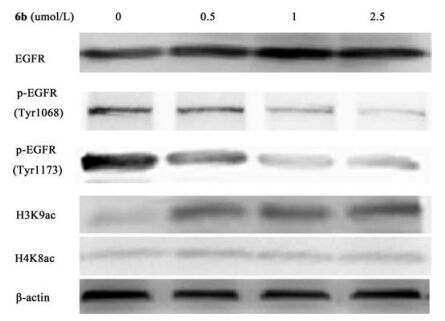
Given the outstanding potency of synthetic hybrids on enzymes and cancer cell lines, we further evaluated whether the compounds could inhibit the phosphorylation of EGFR and induce histone H3 or H4 hyperacetylation on the cellular level. Compound 6b was selected for further experimentation. As shown in Fig. 1, compound 6b suppressed EGFR phosphorylation and up-regulated H3 hyperacetylation in a dose-dependent manner in A549 cells, with marked inhibition of p-EGFR (Tyr1068 and Tyr1173) at 1 μmol/L and obvious up-regulation of histone H3 (Lys9) hyperacetylation at 0.5 μmol/L. However, 6b had minimal effects on histone H4 (Lys8) at the concentration of 2.5 μmol/L. These results further supported the activities of 6b derived from its dual inhibition against EGFR and HDAC.

|
Download:
|
| Fig. 1. Western blot analysis of A549 cells treated for 10 h with compound 6b. | |
{kind=link}
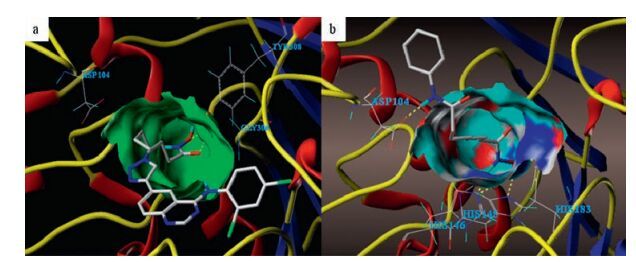
To explore the binding modes of synthetic multitarget compounds with the respective enzymes, compound 6b was docked into active sites of EGFR (PDB ID code: 1XKK [30]) and HDAC2 (PDB ID code: 4LXZ [31]). As depicted in Fig. 2a (compound 6b with EGFR) and Fig. 2b (erlotinib with EGFR), the quinazoline core and 4-aniline group of 6b are oriented in a manner similar to erlotinib. The N1 of quinazoline forms a hydrogen bond with Met793 while the phenyl-amino functional group occupies the hydrophobic pockets of EGFR. C-6 triazole-linked long alkyl chains of hydroxamic acid extend towards the hydrophilic region. Fig. 3a (compound 6b with HDAC2) and Fig. 3b (vorinostat with HDAC2) demonstrates that 6b also mimic the interactions of vorinostat with HDAC2 to quite a high degree. The 4-aminoquinazoline group is found to fit into the surface outside the HDAC active pocket while the hydroxamic acid group extends into the active site and chelates with zinc. The hydroxamic acid group forms two hydrogen bonds with Tyr308. The above docking results may explain why compound 6b displayed the outstanding inhibitory activities against EGFR and HDAC.

|
Download:
|
| Fig. 2. Proposed binding mode of (a) compound 6b and (b) erlotinib with EGFR (1XKK). | |
{kind=link}

|
Download:
|
| Fig. 3. Proposed binding mode of (a) compound 6b and (b) vorinostat with HDAC2 (4LXZ). | |
{kind=link}
3. Conclusion
In conclusion, novel 6-substituted-4-aminoquinazolin derivatives were synthesized and their inhibitory activities against five enzymes (VEGFR2, HER2, EGFR, HDAC1, and HDAC6) and five cell lines (A549, BT-474, A431, SK-BR-3, and NCI-H1975) were evaluated. The introduction of a triazole linked vorinostat-like segment dramatically changed the selectivity profiles of newly synthetic compounds. Among them, compound 6b displayed outstanding potencies against EGFR, HDAC1, and HDAC6 and best anti-proliferative activities on five cancer cell lines. Compound 6b also obviously inhibited the phosphorylation of EGFR and induced histone H3 hyperacetylation on the cellular level. Our study provided evidence that network-based multitarget inhibitor by taking into account the synergy effect of EGFR and HDAC might confer the better benefits than single-targeted inhibitors in cancer therapy. Further modification of the structure of 6b may produce a novel type of anticancer drug candidate, which is in progress.
4. ExperimentalThe synthetic route to compounds 6a-6h is shown in Scheme 2. The keyintermediate 9 wasgeneratedfromcommerciallyavailable compound 7 via cyclization and chlorination [32]. Coupling anilines with 9 produced compounds 10a-10d [32]. Compounds 11a-11d were prepared through Sonogashira reaction and deprotection [33]. The key intermediates 14a and 14b were obtained from commercially available compounds 12a and 12b respectively via azidation and amidation [34]. Coupling 11a-11d [33] with 14a-14b followed by acidification [35] afforded the final products 6a-6h.

|
Download:
|
| Scheme 2. Synthesis of 6a–6h. Reagents and conditions: (a) NH2CHO, reflux, 3h; (b) SOCl2, DMF(cat), reflux, 6h; (c) anilines, 2-propanol, 80 ℃, 6h; (d) (ⅰ) trimethylsilylacetylene, Pd(PPh3)2Cl2, CuI, THF/Et3N, reflux, 12h; (ⅱ) TBAF, THF, r.t., 0.5h (two steps); (e) NaN3, DMF, 80 ℃, 48h; (f) NH2OTHP, EDCI, DMAP, CHCl3, r.t., 12h; (g) CuSO4·5H2O, sodium ascorbate, DMF, 60 ℃, 3h; (h) HCl, dioxane, r.t., 1h. | |
{kind=link}
4.1. General procedure for compounds 10a-10d
To a solution of compound 9 (5.8g, 20mmol) in 2-propanol was added anilines (22mmol) at room temperature (r.t.). Then the reaction mixture was heated to 80 ℃ for 2hours. After the start material was completed, the mixture was filtered through celite, and the cake was washed by 2-propanol, then dried to obtain the desired compounds 10a-10d.
N-(4-Bromo-2-fluorophenyl)-6-iodoquinazolin-4-amine (10a): Yield 96%; mp 240-242 ℃; 1H NMR (400MHz, DMSO-d6): δ 12.02 (s, 1H), 9.35 (d, 1H, J=1.5Hz), 8.94 (s, 1H), 8.40 (dd, 1H, J=8.8, 1.6Hz), 7.84-7.76 (m, 2H), 7.61-7.50 (m, 2H).
6-Iodo-N-(3-methoxyphenyl)quinazolin-4-amine (10b): Yield 92%; mp 286-288 ℃; 1H NMR (400MHz, DMSO-d6): δ 11.76 (s, 1H), 9.42 (d, 1H, J=1.3Hz), 8.95 (s, 1H), 8.36 (dd, 1H, J=8.7, 1.5Hz), 7.80 (d, 1H, J=8.7Hz), 7.42-7.30 (m, 3H), 6.90 (dd, 1H, J=7.3, 1.7Hz), 3.78 (s, 3H).
N-(3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)-6-iodoquinazolin-4-amine (10c): Yield 91%; mp 244-246 ℃; 1H NMR (400MHz, DMSO-d6): δ 11.90 (s, 1H), 9.40 (d, 1H, J=1.3Hz), 8.98 (s, 1H), 8.75 (d, 1H, J=4.5Hz), 8.39 (dd, 1H, J=8.7, 1.6Hz), 8.18 (td, 1H, J=7.8, 1.4Hz), 7.93 (d, 1H, J=2.5Hz), 7.79 (t, 2H, J=9.2Hz), 7.68 (dd, 1H, J=8.9, 2.5Hz), 7.65-7.61 (m, 1H), 7.38 (d, 1H, J=9.0Hz), 5.47 (s, 2H).
N-(3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)-6-iodoquinazolin-4-amine (10d): Yield 94%; mp 279-281 ℃; 1H NMR (400MHz, DMSO-d6): δ 11.86 (s, 1H), 9.40 (s, 1H), 9.05 (s, 1H), 8.40 (d, 1H, J=2.2Hz), 7.87-7.75 (m, 2H), 7.67 (t, 1H, J=8.0Hz), 7.54 (d, 1H, J=7.8Hz), 7.50 (d, 1H, J=8.6Hz), 7.40 (d, 1H, J=8.8Hz), 7.34 (s, 1H), 7.30 (d, 1H, J=8.2Hz).
4.2. General procedure for compounds 11a-11dTo a mixture of 10a or 10d (10mmol), Pd(PPh3)2Cl2 (351mg, 0.5 mmol), and CuI (97mg, 0.5mmol) in THF (30ml) were added trimethylsilylacetylene (2.9mL, 20mmol) and Et3N (4.2mL, 30mmol) under argon atmosphere and the mixture was stirred under reflux condition overnight. After the solvent was removed under reduced pressure, the residue was filtered with short column chromatography on silica gel eluted with dichloromethane/methanol (10/1) to remove the catalysts. The mixture was again concentrated and dissolved in THF (10mL). Tetrabutylammonium fluoride 1.0mol/L solution in THF (10mL, 10mmol) was added and the mixture was stirred for 0.5hour at r.t. After the start material was completed, water was added to quench the reaction, and the mixturewas extracted with ethylacetate, washed by water, saturated NaCl solution and dried by Na2SO4, concentrated and purified by flash silica gel column (CH2Cl2/CH3OH= 50:1, v/v) to obtain desired compounds 11a-11d.
N-(4-Bromo-2-fluorophenyl)-6-ethynylquinazolin-4-amine (11a): Yield 59%; mp 190-192 ℃; 1H NMR (400MHz, DMSO-d6): δ 10.01 (s, 1H), 8.69 (s, 1H), 8.52 (s, 1H), 7.89 (d, 1H, J = 8.5 Hz), 7.78 (d, 1H, J = 8.6 Hz), 7.68 (dd, 1H, J = 9.9, 1.5 Hz), 7.56-7.46 (m, 2H), 4.43 (s, 1H).
6-Ethynyl-N-(3-methoxyphenyl)quinazolin-4-amine (11b): Yield 79%; mp 171-173 ℃; 1H NMR (400 MHz, DMSO-d6): δ 9.85 (s, 1H), 8.81 (s, 1H), 8.63 (s, 1H), 7.87 (dd, 1H, J = 8.5, 1.4 Hz), 7.76 (d, 1H, J = 8.6 Hz), 7.57 (s, 1H), 7.52 (d, 1H, J = 7.9 Hz), 7.30 (t, 1H, J = 8.1 Hz), 6.72 (dd, 1H, J = 8.1, 1.9 Hz), 4.44 (s, 1H), 3.78 (s, 3H).
N-(3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)-6-ethynylquinazolin-4-amine (11c): Yield 44%; mp 197-199 ℃; 1H NMR (400 MHz, DMSO-d6): δ 9.87 (s, 1H), 8.74 (d, 1H, J = 1.0 Hz), 8.59 (d, 2H, J = 5.1 Hz), 8.06 (d, 1H, J = 2.5 Hz), 7.90-7.84 (m, 2H), 7.75 (d, 1H, J = 2.0 Hz), 7.73 (d, 1H, J = 2.5 Hz), 7.58 (d, 1H, J = 7.8 Hz), 7.36 (dd, 1H, J = 6.9, 5.1 Hz), 7.26 (d, 1H, J = 9.0 Hz), 5.29 (s, 2H), 4.41 (s, 1H).
N-(3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)-6-ethynylquinazolin-4-amine (11d): Yield 38%; mp 169-171 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.03 (s, 1H), 8.80 (d, 1H, J = 1.1 Hz), 8.69 (s, 1H), 8.32 (d, 1H, J = 2.5 Hz), 7.95 (dd, 1H, J = 8.9, 2.6 Hz), 7.89 (dd, 1H, J = 8.6, 1.5 Hz), 7.79 (d, 1H, J = 8.6 Hz), 7.62 (t, 1H, J = 8.0 Hz), 7.48 (d, 1H, J = 7.8 Hz), 7.37-7.33 (m, 1H), 7.26 (s, 1H), 7.22 (dd, 1H, J = 8.2, 2.4 Hz), 4.45 (s, 1H).
4.3. General procedure for compounds 13a and 13bTo a solution of compound 12a or 12b (20 mol) in DMF (50 mL) was added sodium azide (3.9 g, 60 mmol) at r.t. Then the reaction mixture was heated to 80 ℃ for 48 hours. After the start material was completed, water was added and the desired product was extracted with ethyl acetate, washed by water, saturated NaCl solution and dried by Na2SO4, concentrated and to obtain desired compounds 13a and 13b.
6-Azidohexanoic acid (13a): Yield 86%; 1H NMR (400 MHz, CDCl3): δ 11.20 (s, 1H), 3.29 (t, 2H, J = 6.9 Hz), 2.38 (t, 2H, J = 7.4 Hz), 1.73-1.58 (m, 4H), 1.48-1.39 (m, 2H).
7-Azidoheptanoic acid (13b): Yield 82%; 1H NMR (400 MHz, CDCl3): δ 11.36 (s, 1H), 3.27 (t, 2H, J = 6.9 Hz), 2.36 (t, 2H, J = 7.4 Hz), 1.70-1.56 (m, 4H), 1.45-1.34 (m, 4H).
4.4. General procedure for compounds 14a and 14bTo a mixture 13a or 13b (10 mmol) and O-(tetrahydro-2H-pyran-2-yl)hydroxylamine (1.29 g, 11 mmol) in chloroform (30 mL) was added EDCI (2.52 g, 13.2 mmol) followed by DMAP (122 mg, 1 mmol). The resulting mixture was stirred overnight at RT. After the start material was completed, the reaction mixture was concentrated, and water was added. Then the mixture was extracted with CHCl3, washed by saturated NaCl solution and dried by Na2SO4, concentrated and purified by flash silica gel column (PE/CH2Cl2 = 1:1, v/v) to obtain desired compounds 14a and 14b.
6-Azido-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (14a): Yield 91%; 1H NMR (400 MHz, CDCl3): δ 8.38 (s, 1H), 4.94 (s, 1H), 3.95 (d, 1H, J = 9.0 Hz), 3.63 (m, 1H), 3.27 (t, 2H, J = 6.8 Hz), 2.14-1.76 (m, 4H), 1.74-1.50 (m, 8H), 1.47-1.35 (m, 2H).
7-Azido-N-((tetrahydro-2H-pyran-2-yl)oxy)heptanamide (14b): Yield 90%; 1H NMR (400 MHz, CDCl3): δ 8.76 (s, 1H), 4.89 (s, 1H), 3.95 (d, 1H, J = 9.4 Hz), 3.63 (m, 1H), 3.26 (t, 3H, J = 6.9 Hz), 2.19-1.76 (m, 4H), 1.73-1.48 (m, 8H), 1.45-1.29 (m, 4H).
4.5. General procedure for compounds 15a-15hTo a solution of 11a or 11b (2 mmol) and 14a or 14b (2 mmol) in DMF (5 mL) were added CuSO4·5H2O (0.15 g, 0.6 mmol) and sodium ascorbate (0.2 g, 1 mmol). Then the mixture was heated at 70 ℃ for 3 hours. After the start material was completed, water was added and the mixture was extracted with ethyl acetate, washed by water, saturated NaCl solution and dried by Na2SO4, concentrated and purified by flash silica gel column (PE/THF = 1:1, v/v) to obtain desired compounds 15a-15h.
6-(4-(4-((4-Bromo-2-fluorophenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (15a): Yield 72%; mp 100-102 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.93 (s, 1H), 10.12 (s, 1H), 9.01 (s, 1H), 8.66 (s, 1H), 8.51 (s, 1H), 8.31 (d, 1H, J = 8.4 Hz), 7.88 (d, 1H, J = 8.2 Hz), 7.69 (d, 1H, J = 9.6 Hz), 7.61-7.52 (m, 1H), 7.50 (d, 1H, J = 7.8 Hz), 4.77 (s, 1H), 4.47 (t, 2H, J = 6.8 Hz), 3.95-3.77 (m, 1H), 3.47 (d, 1H, J = 11.0 Hz), 2.10-1.78 (m, 4H), 1.75-1.38 (m, 8H), 1.34-1.14 (m, 2H).
7-(4-(4-((4-Bromo-2-fluorophenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2H-pyran-2-yl)oxy)heptanamide (15b): Yield 58%; mp 134-136 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.92 (s, 1H), 10.11 (s, 1H), 9.01 (s, 1H), 8.67 (s, 1H), 8.51 (s, 1H), 8.31 (d, 1H, J = 8.5 Hz), 7.88 (d, 1H, J = 8.5 Hz), 7.69 (d, 1H, J = 9.8 Hz), 7.61-7.53 (m, 1H), 7.49 (d, 1H, J = 8.0 Hz), 4.80 (s, 1H), 4.46 (t, 2H, J = 6.8 Hz), 3.97-3.83 (m, 1H), 3.48 (d, 1H, J = 11.3 Hz), 2.12-1.80 (m, 4H), 1.76-1.40 (m, 8H), 1.38-1.20 (m, 4H).
6-(4-(4-((3-Methoxyphenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (15c): Yield 46%; mp 79-81 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.95 (s, 1H), 9.97 (s, 1H), 9.07 (s, 1H), 8.67 (s, 1H), 8.62 (s, 1H), 8.30 (d, 1H, J = 8.6 Hz), 7.87 (d, 1H, J = 8.6 Hz), 7.58 (s, 1H), 7.54 (d, 1H, J = 7.9 Hz), 7.32 (t, 1H, J = 8.1 Hz), 6.74 (dd, 1H, J = 8.2, 2.2 Hz), 4.77 (s, 1H), 4.48 (t, 2H, J = 6.5 Hz), 3.95-3.78 (m, 1H), 3.81 (s, 3H), 3.46 (d, 1H, J = 10.9 Hz), 2.05-1.83 (m, 4H), 1.70-1.40 (m, 8H), 1.32-1.20 (m, 2H).
7-(4-(4-((3-Methoxyphenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2H-pyran-2-yl)oxy)heptanamide (15d): Yield 62%; mp 151-153 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.93 (s, 1H), 9.97 (s, 1H), 9.07 (s, 1H), 8.68 (s, 1H), 8.62 (s, 1H), 8.30 (dd, 1H, J = 8.6, 1.4 Hz), 7.87 (d, 1H, J = 8.6 Hz), 7.58 (s, 1H), 7.53 (d, 1H, J = 8.0 Hz), 7.32 (t, 1H, J = 8.1 Hz), 6.74 (dd, 1H, J = 8.2, 2.2 Hz), 4.80 (s, 1H), 4.47 (t, 2H, J = 6.9 Hz), 4.00-3.80 (m, 1H), 3.80 (s, 3H), 3.49 (d, 1H, J = 11.2 Hz), 2.10-1.78 (m, 4H), 1.72-1.40 (m, 8H), 1.36-1.18 (m, 4H).
6-(4-(4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino) quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (15e): Yield 43%; mp 122-124 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.90 (s, 1H), 10.00 (s, 1H), 9.03 (s, 1H), 8.64 (s, 1H), 8.62-8.59 (m, 1H), 8.58 (s, 1H), 8.27 (dd, 1H, J = 8.6, 1.6 Hz), 8.06 (d, 1H, J = 2.5 Hz), 7.88 (m, 2H), 7.77 (dd, 1H, J = 9.0, 2.5 Hz), 7.60 (d, 1H, J = 7.8 Hz), 7.37 (dd, 1H, J = 7.0, 5.4 Hz), 7.29 (d, 1H, J = 9.1 Hz), 5.30 (s, 2H), 4.76 (s, 1H), 4.46 (t, 2H, J = 6.9 Hz), 3.95-3.83 (m, 1H), 3.48 (d, 1H, J = 11.2 Hz), 2.06-1.80 (m, 4H), 1.73-1.40 (m, 8H), 1.35-1.20 (m, 2H).
7-(4-(4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino) quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2H-pyran-2-yl)oxy)heptanamide (15f): Yield 34%; mp 168-170 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.89 (s, 1H), 9.99 (s, 1H), 9.03 (s, 1H), 8.65 (s, 1H), 8.60 (d, 1H, J = 4.2 Hz), 8.58 (s, 1H), 8.27 (dd, 1H, J = 8.6, 1.6 Hz), 8.06 (d, 1H, J = 2.5 Hz), 7.88 (m, 2H), 7.77 (dd, 1H, J = 9.0, 2.5 Hz), 7.60 (d, 1H, J = 7.8 Hz), 7.37 (dd, 1H, J = 6.7, 5.0 Hz), 7.29 (d, 1H, J = 9.1 Hz), 5.30 (s, 2H), 4.79 (s, 1H), 4.46 (t, 2H, J = 6.9 Hz), 3.94-3.80 (m, 1H), 3.46 (d, 1H, J = 11.1 Hz), 2.05-1.83 (m, 4H), 1.70-1.40 (m, 8H), 1.32-1.21 (m, 4H).
6-(4-(4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl) amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (15g): Yield 51%; mp 119-121 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.93 (s, 1H), 10.21 (s, 1H), 9.10 (s, 1H), 8.70 (s, 1H), 8.68 (s, 1H), 8.32 (dd, 2H, J = 9.3, 5.4 Hz), 8.00 (d, 1H, J = 6.7 Hz), 7.90 (d, 1H, J = 8.6 Hz), 7.64 (t, 1H, J = 8.0 Hz), 7.49 (d, 1H, J = 7.7 Hz), 7.37 (d, 1H, J = 8.9 Hz), 7.29-7.22 (m, 2H), 4.77 (s, 1H), 4.47 (t, 2H, J = 6.7 Hz), 3.96-3.83 (m, 1H), 3.46 (d, 1H, J = 11.4 Hz), 2.05-1.85 (m, 4H), 1.70-1.41 (m, 8H), 1.31-1.22 (m, 2H).
7-(4-(4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl) amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-((tetrahydro-2Hpyran-2-yl)oxy)heptanamide (15h): Yield 45%; mp 175-177 ℃; 1H NMR (400 MHz, DMSO-d6): δ 10.90 (s, 1H), 10.19 (s, 1H), 9.09 (s, 1H), 8.69 (s, 1H), 8.68 (s, 1H), 8.31 (d, 2H, J = 9.9 Hz), 7.99 (dd, 1H, J = 8.9, 2.2 Hz), 7.90 (d, 1H, J = 8.7 Hz), 7.64 (t, 1H, J = 8.0 Hz), 7.49 (d, 1H, J = 7.8 Hz), 7.37 (d, 1H, J = 8.9 Hz), 7.30-7.21 (m, 2H), 4.79 (s, 1H), 4.47 (t, 2H, J = 7.0 Hz), 3.96-3.86 (m, 1H), 3.48 (d, 1H, J = 11.4 Hz), 2.03-1.85 (m, 4H), 1.72-1.40 (m, 8H), 1.33-1.23 (m, 4H).
4.6. General procedure for compounds 6a-6h2.0 mol/L HCl in dioxane (1 mL, 2 mmol) was added dropwise to a solution of the 15a-15h intermediate (0.5 mmol) in dioxane (5 mL) and the mixture was stirred at r.t. for 1 h. The precipitate was filtered off, washed with ethyl ether, and dried to give the desired hydroxamic acid 6a-6h.
6-(4-(4-((4-Bromo-2-fluorophenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-hydroxyhexanamide (6a): Yield 94%; mp 213-215 ℃; 1H NMR (400 MHz, DMSO-d6): δ 12.15 (s, 1H), 10.41 (s, 1H), 9.54 (s, 1H), 8.93 (s, 1H), 8.88 (s, 1H), 8.59 (dd, 1H, J = 8.7, 1.3 Hz), 8.08 (d, 1H, J = 8.7 Hz), 7.86-7.78 (m, 1H), 7.65-7.48 (m, 2H), 4.47 (t, 2H, J = 6.9 Hz), 2.04-1.78 (m, 4H), 1.62-1.46 (m, 2H), 1.36-1.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 169.39, 161.15, 157.21 (d, J = 249.5 Hz), 151.46, 145.30, 139.03, 133.70, 131.79, 130.62, 128.53 (d, J = 3.0 Hz), 124.47 (d, J = 12.1 Hz), 123.29, 121.49, 121.16, 121.01, 120.30 (d, J = 24.2 Hz), 114.29, 50.17, 32.52, 29.81, 25.92, 24.97. HR-MS (ESI) m/z: Calcd. for [M + H]+ 514.1002; Found: 514.0994.
7-(4-(4-((4-Bromo-2-fluorophenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-hydroxyheptanamide (6b): Yield 97%; mp 199-201 ℃; 1H NMR (400 MHz, DMSO-d6): δ 12.20 (s, 1H), 10.40 (s, 1H), 9.57 (s, 1H), 8.92 (s, 1H), 8.88 (s, 1H), 8.60 (dd, 1H, J = 8.7, 1.3 Hz), 8.08 (d, 1H, J = 8.7 Hz), 7.87-7.78 (m, 1H), 7.66-7.49 (m, 2H), 4.47 (t, 2H, J = 6.9 Hz), 2.00-1.77 (m, 4H), 1.55-1.38 (m, 2H), 1.37-1.18 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 169.53, 161.16, 157.21 (d, J = 249.5 Hz), 151.41, 145.29, 138.90, 133.69, 131.81, 130.63, 128.52 (d, J = 4.0 Hz), 124.45 (d, J = 13.1 Hz), 123.29, 121.38, 121.11, 121.02, 120.30 (d, J = 23.2 Hz), 114.27, 50.25, 32.63, 29.99, 28.44, 26.06, 25.40. HR-MS (ESI) m/z: Calcd. for [M + H]+ 528.1159; Found: 528.1158.
N-Hydroxy-6-(4-(4-((3-methoxyphenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)hexanamide (6c): Yield 74%; mp 229-231 ℃; 1H NMR (400 MHz, DMSO-d6): δ 11.94 (s, 1H), 10.45 (s, 1H), 9.63 (s, 1H), 8.98 (s, 1H), 8.94 (s, 1H), 8.56 (d, 1H, J = 8.8 Hz), 8.04 (d, 1H, J = 8.7 Hz), 7.49-7.27 (m, 3H), 7.00-6.81 (m, 1H), 4.46 (t, 2H, J = 6.8 Hz), 3.80 (s, 3H), 2.04-1.90 (m, 4H), 1.63-1.45 (m, 2H), 1.36-1.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 169.42, 160.21, 159.86, 151.00, 145.38, 138.32, 138.27, 133.28, 131.66, 129.96, 123.42, 121.31, 120.84, 117.41, 114.59, 112.54, 111.30, 55.82, 50.15, 32.52, 29.77, 25.91, 24.97. HR-MS (ESI) m/z: Calcd. for [M + H]+ 448.2097; Found: 448.2100.
N-Hydroxy-7-(4-(4-((3-methoxyphenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)heptanamide (6d): Yield 93%; mp 164-166 ℃; 1H NMR (400 MHz, DMSO-d6): δ 11.89 (s, 1H), 10.40 (s, 1H), 9.60 (s, 1H), 8.95 (s, 1H), 8.94 (s, 1H), 8.56 (d, 1H, J = 8.7 Hz), 8.03 (d, 1H, J = 8.7 Hz), 7.52-7.29 (m, 3H), 6.93 (dd, 1H, J = 7.5, 4.0 Hz), 4.46 (t, 2H, J = 6.9 Hz), 3.80 (s, 3H), 2.00-1.77 (m, 4H), 1.57-1.40 (m, 2H), 1.38-1.17 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 169.53, 160.19, 159.88, 151.18, 145.40, 138.70, 138.34, 133.30, 131.62, 129.99, 123.34, 121.18, 121.13, 117.37, 114.66, 112.49, 111.25, 55.82, 50.23, 32.63, 29.98, 28.43, 26.07, 25.40. HR-MS (ESI) m/z: Calcd. for [M + H]+ 462.2254; Found: 462.2246.
6-(4-(4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino) quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-hydroxyhexanamide (6e): Yield 88%; mp 213-215 ℃; 1H NMR (400 MHz, DMSO-d6): δ 12.17 (s, 1H), 10.32 (s, 1H), 9.72 (s, 1H), 9.00 (s, 1H), 8.96 (s, 1H), 8.80 (d, 1H, J = 4.9 Hz), 8.59 (d, 1H, J = 8.6 Hz), 8.27 (t, 1H, J = 7.7 Hz), 8.07 (d, 1H, J = 8.7 Hz), 8.03 (s, 1H), 7.89 (d, 1H, J = 7.8 Hz), 7.79 (d, 1H, J = 9.0 Hz), 7.75-7.67 (m, 1H), 7.40 (d, 1H, J = 9.0 Hz), 5.43 (s, 2H), 4.47 (t, 2H, J = 6.5 Hz), 2.27-1.77 (m, 4H), 1.62-1.40 (m, 2H), 1.38-1.05 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 174.85, 160.15, 154.33, 152.03, 150.96, 146.83, 145.32, 141.62, 137.89, 133.33, 131.69, 131.18, 126.96, 125.29, 125.09, 123.72, 123.45, 121.54, 121.35, 120.58, 114.59, 114.53, 69.66, 50.16, 33.94, 29.86, 25.91, 24.39. HRMS (ESI) m/z: Calcd. for [M + H]+ 559.1973; Found: 559.1966.
7-(4-(4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino) quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-hydroxyheptanamide (6f): Yield 83%; mp 200-201 ℃; 1H NMR (400 MHz, DMSO-d6): δ 12.10 (s, 1H), 10.42 (s, 1H), 9.66 (s, 1H), 8.97 (s, 1H), 8.97 (s, 1H), 8.72 (d, 1H, J = 4.7 Hz), 8.57 (d, 1H, J = 8.6 Hz), 8.11 (t, 1H, J = 7.7 Hz), 8.04 (d, 1H, J = 8.7 Hz), 8.01 (d, 1H, J = 2.4 Hz), 7.78 (s, 1H), 7.75 (s, 1H), 7.62-7.54 (m, 1H), 7.38 (d, 1H, J = 9.1 Hz), 5.44 (s, 2H), 4.46 (t, 2H, J = 6.9 Hz), 2.05-1.78 (m, 4H), 1.57-1.38 (m, 2H), 1.36-1.15 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 169.54, 160.15, 155.13, 152.17, 151.04, 147.86, 145.33, 140.24, 137.95, 133.38, 131.71, 131.03, 126.94, 125.27, 124.58, 123.41, 123.10, 121.52, 121.28, 120.65, 114.56, 70.36, 50.23, 32.62, 29.98, 28.43, 26.06, 25.41. HR-MS (ESI) m/z: Calcd. for [M + H]+ 573.2129; Found: 573.2114.
6-(4-(4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-hydroxyhexanamide (6g): Yield 65%; mp 233-235 ℃; 1H NMR (400 MHz, DMSO-d6): δ 12.02 (s, 1H), 10.40 (s, 1H), 9.62 (s, 1H), 8.99 (s, 1H), 8.94 (s, 1H), 8.54 (d, 1H, J = 8.6 Hz), 8.20 (s, 1H), 8.02 (d, 1H, J = 8.7 Hz), 7.89 (d, 1H, J = 8.7 Hz), 7.71-7.54 (m, 1H), 7.51 (d, 1H, J = 7.7 Hz), 7.36 (t, 1H, J = 9.5 Hz), 7.31 (s, 1H), 7.26 (d, 1H, J = 8.3 Hz), 4.44 (t, 2H, J = 6.5 Hz), 2.01-1.80 (m, 4H), 1.60-1.45 (m, 2H), 1.34-1.15 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 169.40, 160.15, 157.52, 151.34, 148.76, 145.36, 135.14, 133.40, 132.11, 131.67, 131.47, 131.15, 127.03, 125.55, 125.26, 123.41, 122.81, 122.48, 121.36, 121.12, 120.57, 114.75, 114.08, 50.15, 32.51, 29.79, 25.91, 24.98. HR-MS (ESI) m/z: Calcd. for [M + H]+ 612.1738; Found: 612.1726.
7-(4-(4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl) amino)quinazolin-6-yl)-1H-1, 2, 3-triazol-1-yl)-N-hydroxyheptanamide (6h): Yield 47%; mp 255-257 ℃; 1H NMR (400 MHz, DMSO-d6): δ 12.23 (s, 1H), 10.31 (s, 1H), 9.73 (s, 1H), 9.03 (s, 1H), 9.00 (s, 1H), 8.59 (d, 1H, J = 8.8 Hz), 8.22 (d, 1H, J = 2.4 Hz), 8.08 (d, 1H, J = 8.7 Hz), 7.91 (dd, 1H, J = 8.8, 2.4 Hz), 7.66 (t, 1H, J = 8.0 Hz), 7.53 (d, 1H, J = 7.7 Hz), 7.40 (d, 1H, J = 8.8 Hz), 7.34 (s, 1H), 7.28 (d, 1H, J = 8.2 Hz), 4.46 (t, 2H, J = 6.9 Hz), 2.29-1.77 (m, 4H), 1.57-1.42 (m, 2H), 1.38-1.20 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 174.94, 160.34, 157.48, 151.06, 148.95, 145.30, 138.09, 134.93, 133.52, 132.13, 131.82, 131.16, 127.26, 125.78, 125.24, 123.50, 122.44, 121.39, 120.73, 120.56, 114.65, 114.15, 50.23, 34.02, 29.96, 28.39, 26.06, 24.76. HRMS (ESI) m/z: Calcd. for [M + H]+ 626.1894; Found: 626.1884.
Kinase inhibition assay, HDAC inhibition assay, cell culture, cytotoxicity assay, western blot methods, docking methods, 1H NMR and 13C NMR spectra, and high resolution mass spectrometry (HRMS) of final compounds 6a-6h can be found in supporting information.
AcknowledgmentThis work is supported by funding from the National Natural Science Foundation of China (No. 21272134) and Shenzhen Municipal Government (No. CXB201104210014A and 20150113A0410006).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.01.003.
| [1] | R. Taby, J.P.J. Issa. Cancer epigenetics. CA Cancer J. Clin. 60(2010)376–392. DOI:10.3322/caac.20085 |
| [2] | B.S. Mann, J.R. Johnson, M.H. Cohen, R. Justice, R. Pazdur. FDA approval summary:vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12(2007)1247–1252. DOI:10.1634/theoncologist.12-10-1247 |
| [3] | L. Ellis, Y. Pan, G.K. Smyth, et al., Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin. Cancer Res. 14(2008)4500–4510. DOI:10.1158/1078-0432.CCR-07-4262 |
| [4] | C. Grant, F. Rahman, R. Piekarz, et al., Romidepsin:a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 10(2010)997–1008. DOI:10.1586/era.10.88 |
| [5] | C. Yu, B.B. Friday, J.P. Lai, et al., Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin. Cancer Res. 13(2007)1140–1148. DOI:10.1158/1078-0432.CCR-06-1751 |
| [6] | E.P. Jane, D.R. Premkumar, S.O. Addo-Yobo, I.F. Pollack. Abrogation of mitogenactivated protein kinase and Akt signaling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human Glioma cells. J. Pharmacol. Exp. Ther. 331(2009)327–337. DOI:10.1124/jpet.109.155705 |
| [7] | S. Mahboobi, S. Dove, A. Sellmer, et al., Design of chimeric histone deacetylaseand tyrosine kinase-inhibitors:a series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rb, and histone deacetylases. J. Med. Chem. 52(2009)2265–2279. DOI:10.1021/jm800988r |
| [8] | X. Cai, H.X. Zhai, J. Wang, et al., Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 53(2010)2000–2009. DOI:10.1021/jm901453q |
| [9] | S. Mahboobi, A. Sellmer, M. Winkler, et al., Novel chimeric histone deacetylase inhibitors:a series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2(HER2), and histone deacetylase activity,. J. Med. Chem. 53(2010)8546–8555. DOI:10.1021/jm100665z |
| [10] | Y. Li, C. Tan, C. Gao, et al., Discovery of benzimidazole derivatives as novel multi-target EGFR, VEGFR-2 and PDGFR kinase inhibitors. Bioorg. Med. Chem. 19(2011)4529–4535. DOI:10.1016/j.bmc.2011.06.022 |
| [11] | X. Luan, C. Gao, N. Zhang, et al., Exploration of acridine scaffold as a potentially interesting scaffold for discovering novel multi-target VEGFR-2 and Src kinase inhibitors. Bioorg. Med. Chem. 19(2011)3312–3319. DOI:10.1016/j.bmc.2011.04.053 |
| [12] | C. Zhang, C. Tan, X. Zu, et al., Exploration of (S)-3-aminopyrrolidine as a potentially interesting scaffold for discovery of novel Abl and PI3 K dual inhibitors. Eur. J. Med. Chem. 46(2011)1404–1414. DOI:10.1016/j.ejmech.2011.01.020 |
| [13] | F. Jin, D. Gao, Q. Wu, et al., Exploration of N-(2-aminoethyl)piperidine-4-carboxamide as a potential scaffold for development of VEGFR-2, ERK-2 and Abl-1 multikinase inhibitor. Bioorg. Med. Chem. 21(2013)5694–5706. DOI:10.1016/j.bmc.2013.07.026 |
| [14] | F. Jin, D. Gao, C. Zhang, et al., Exploration of 1-(3-chloro-4-(4-oxo-4Hchromen-2-yl)phenyl)-3-phenylurea derivatives as selective dual inhibitors of Raf1 and JNK1 kinases for anti-tumor treatment. Bioorg. Med. Chem. 21(2013)824–831. DOI:10.1016/j.bmc.2012.04.006 |
| [15] | X.L. Lang, Q.S. Sun, Y.Z. Chen, et al., Novel synthetic 9-benzyloxyacridine analogue as both tyrosine kinase and topoisomerase I inhibitor. Chin. Chem. Lett. 24(2013)677–680. DOI:10.1016/j.cclet.2013.05.018 |
| [16] | Z. Cui, X. Li, L. Li, et al., Design, synthesis and evaluation of acridine derivatives as multi-target Src and MEK kinase inhibitors for anti-tumor treatment. Bioorg. Med. Chem. 24(2016)261–269. DOI:10.1016/j.bmc.2015.12.011 |
| [17] | L. Li, C.L. Zhang, H.R. Song, et al., Discovery of novel dual inhibitors of VEGFR and PI3 K kinases containing 2-ureidothiazole scaffold. Chin. Chem. Lett. 27(2016)1–6. DOI:10.1016/j.cclet.2015.09.008 |
| [18] | W. Zhang, B. Zhang, W. Zhang, et al., Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNAbinding ligands and topoisomerase Ⅱ inhibitors. Eur. J. Med. Chem. 116(2016)59–70. DOI:10.1016/j.ejmech.2016.03.066 |
| [19] | J.J.L. Liao. Molecular recognition of protein kinase binding pockets fordesign of potent and selective kinase inhibitors. J. Med. Chem. 50(2007)409–424. DOI:10.1021/jm0608107 |
| [20] | M. Cai, J. Hu, J.L. Tian, et al., Novel hybrids fromN-hydroxyarylamideand indole ring through click chemistry as histone deacetylase inhibitors with potent antitumor activities. Chin. Chem. Lett. 26(2015)675–680. DOI:10.1016/j.cclet.2015.03.015 |
| [21] | S.K. Bhattacharya, E.D. Cox, J.C. Kath, et al., Achieving selectivity between highly homologous tyrosine kinases:a novel selective erbB2 inhibitor. Biochem. Biophys. Res. Commun. 307(2003)267–273. DOI:10.1016/S0006-291X(03)01160-4 |
| [22] | P. Bali, M. Pranpat, R. Swaby, et al., Activity of suberoylanilide hydroxamic acid against human breastcancercells with amplification ofHer-2. Clin. Cancer Res. 11(2005)6382–6389. DOI:10.1158/1078-0432.CCR-05-0344 |
| [23] | B.T. Scroggins, K. Robzyk, D. Wang, et al., An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25(2007)151–159. DOI:10.1016/j.molcel.2006.12.008 |
| [24] | P. Steiner, C. Joynes, R. Bassi, et al., Tumor growth inhibition with cetuximab and chemotherapy in non-small cell lung cancer xenografts expressing wildtype and mutated epidermal growth factor receptor. Clin. Cancer Res. 13(2007)1540–1551. DOI:10.1158/1078-0432.CCR-06-1887 |
| [25] | S. Chang, L. Zhang, S. Xu, et al., Design, synthesis, and biological evaluation of novel conformationally constrained inhibitors targeting epidermal growth factor receptor threonine790!methionine790 mutant,. J. Med. Chem. 55(2012)2711–2723. DOI:10.1021/jm201591k |
| [26] | B. Chu, F. Liu, L. Li, et al., A benzimidazole derivative exhibiting antitumor activity blocks EGFR and HER2 activity and upregulates DR5 in breast cancer cells. Cell Death Dis. 6(2015)e1686. DOI:10.1038/cddis.2015.25 |
| [27] | Z. Xing, X. Tang, Y. Gao, et al., The human LIS1 is downregulated in hepatocellular carcinoma and plays a tumor suppressor function. Biochem. Biophys. Res. Commun. 409(2011)193–199. DOI:10.1016/j.bbrc.2011.04.117 |
| [28] | G. Merlino, Y. Xu, S. Ishii, et al., Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science 224(1984)417–419. DOI:10.1126/science.6200934 |
| [29] | G.E. Konecny, M.D. Pegram, N. Venkatesan, et al., Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 66(2006)1630–1639. DOI:10.1158/0008-5472.CAN-05-1182 |
| [30] | E.R. Wood, A.T. Truesdale, O.B. McDonald, et al., A unique structure for epidermal growth factor receptor bound to GW572016(Lapatinib):relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 64(2004)6652–6659. DOI:10.1158/0008-5472.CAN-04-1168 |
| [31] | B.E.L. Lauffer, R. Mintzer, R. Fong, et al., Histone deacetylase (hdac) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 288(2013)26926–26943. DOI:10.1074/jbc.M113.490706 |
| [32] | G. Patel, C.E. Karver, R. Behera, et al., Kinase scaffold repurposing for neglected disease drug discovery:discovery of an efficacious lapatanib-derived lead compound for trypanosomiasis. J. Med. Chem. 56(2013)3820–3832. DOI:10.1021/jm400349k |
| [33] | G. Gu, H. Wang, P. Liu, et al., Discovery and structural insight of a highly selective protein kinase inhibitor hit through click chemistry. Chem. Commun. 48(2012)2788–2790. DOI:10.1039/c1cc15851a |
| [34] | J.B. Chen, T.R. Chern, T.T. Wei, et al., Design and synthesis of dual-action inhibitors targeting histone deacetylases and 3-hydroxy-3-methylglutaryl coenzyme a reductase for cancer treatment. J. Med. Chem. 56(2013)3645–3655. DOI:10.1021/jm400179b |
| [35] | V. Hugenberg, B. Riemann, S. Hermann, et al., Inverse 12, 3-Triazole-1-yl-ethyl substituted hydroxamates as highly potent matrix metalloproteinase inhibitors:(radio)synthesis, in vitro and first in vivo evaluation. J. Med. Chem. 56(2013)6858–6870. DOI:10.1021/jm4006753 |