 2017, Vol. 28
2017, Vol. 28 



Chemical investigations have revealed that Penicillium spp. are proficient in using different synthases, like polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs), to creat a vast variety of secondary metabolites [1-5]. Various metabolites with wide ranges of bioactivties, such as anticancer, antimicrobial, anti-inflammatory and insecticidal were isolated from the culture of Penicillium spp. [6-9]. It makes the Penicillium genus a rich and hot source of natural products. P. brefeldianum is one of the well kown Penicillium sp. Its famous product is the 16-membered macrolide antibiotic brefeldin A, an intracellular protein transport inhibitor that also can lead to differentiation and apoptosis of a wide variety of human cancer cells [10, 11]. Recently, during the investigation of an endophytic P. brefeldianum XMK-2 from the rhizome of Pinellia ternata, we isolated one new indoloditerpene, 6, 7-dehydropaxilline (1), one new indole-diketopiperazine, spirotryprostatin F (2), and one new 13-membered macrolide, N-demethylmelearoride A (3), together with 8 known indoloditerpene and indole-diketopiperazine alkaloids (Fig. 1). To the best of our knowledge, the indoloditerpenes and the 13-membered macrolide were reported from this species for the fist time. Meanwhile, when evaluating their bioactivities, these metabolites showed different cytotoxic activities against three human tumor cell lines. The new compounds 2 and 3 showed moderate cytotoxicities against HepG2 and MDA-MB-231 cells, and the known 11 was the most effective one that comparable to the positive control cisplatin. Herein, the details of isolation and structural elucidation of 1-3 are presented, in addition with the cytotoxic evaluation of all the isolates.

|
Download:
|
| Fig. 1. Structures of compounds 1-11. | |
2. Results and discussion
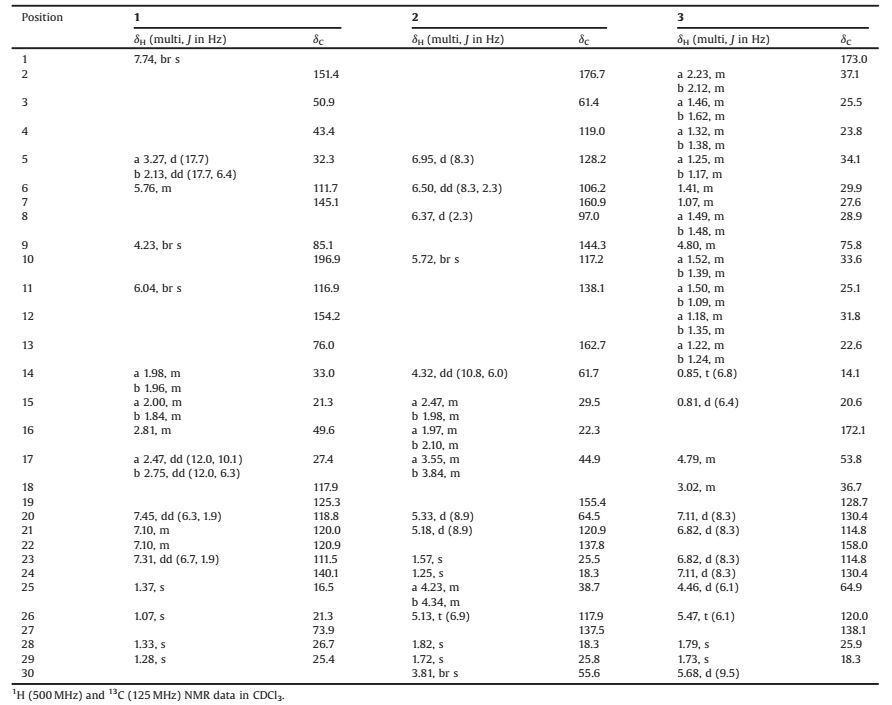
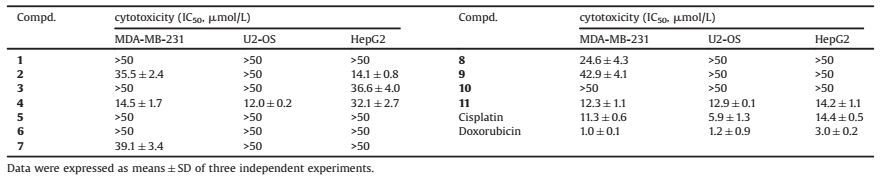
Compound 1 was obstained as a yellow powder. Its molecular formula C27H31NO4 was determined by the positive HRESIMS ion at m/z 434.2325 [M+H]+ (calcd.for C27H32NO4, 434.2326), revealing 13 degrees of unsaturation. The ESIMS fragment ion at m/z 130 [(C9H8N)+], as well as the characteristic UV absorptions at 229 and 277 nm, indicated the presence of an indole moiety in 1 [12, 13]. Its 1H NMR data (Table 1) also displayed the diagnostic indole singals of the NH at δH 7.74 (br s) and the four aromatic protons at δH 7.45 (dd, 1H, J = 6.3, 1.9 Hz, H-20), 7.31 (dd, 1H, J = 6.7, 1.9 Hz, H-23), and 7.10 (m, 2H, H-21, 22). The remaining protons were two olefinic signals at δH 6.04 (br s, 1H, H-11) and 5.76 (m, 1H, H-6), four methyl singlets at δH 1.37 (s, 3H, H3-25), 1.33 (s, 3H, H3-28), 1.28 (s, 3H, H3-29), and 1.07 (s, 3H, H3-26), four methylenes at δH 3.27 (d, 1H, J = 17.7 Hz, H-5a)/2.13 (dd, 1H, J = 17.7, 6.4 Hz, H-5b), 2.75 (dd, 1H, J = 12.0, 6.3 Hz, H-17b)/2.47 (dd, 1H, J = 12.0, 10.1 Hz, H-17a), 2.00 (m, 1H, H-15a)/1.84 (m, 1H, H-15b), and 1.98 (m, 1H, H-14a)/1.96 (m, 1H, H-14b), and two methines at δH 4.23 (br s, 1H, H-9) and 2.81 (m, 1H, H-16). Based on the HSQC spectrum, all the above corresponding carbons could be found in its 13C NMR spectrum (Table 1), which also showed additional resonances of one carbonyl at δC 196.9 and two oxygenated quaternary carbons at δC 76.0 and 73.9. These functional groups accounted for 9 indices of hydrogen deficiency, and the remaining four ones implied other tetracyclic rings in 1. Further NMR data comparison with those of 13-desoxypaxilline (5) revealed the same carbon skeleton of them, which was confimred by the HMBC correlations (Fig. 2). Differently, the HMBC correlations between H-11/H3-26/H-5b and C-13 (δC 76.0) revealed the hydroxylation of C-13. The HMBC correlations from H-6 to C-4/C-7/C-12, from H2-5 to C-6/C-7, and from H-11 to C-7 also established the double bond at Δ6(7). Therefore, its planar structure was identified as 6, 7-dehydropaxilline.
|
|
Table 1 1H NMR and 13C NMR spectroscopic data of compounds 1-3. |

|
Download:
|
| Fig. 2. The key HMBC and ROESY correlations of 1. | |
The relative configuration of 1 was established by ROESY experiment (Fig. 2). The correlations of H3-26/H-14b/H-16 indicated that they were on the same face of the chair form of ring D. The ROESY correlations between H3-25 and H-15a/H-14a/ H-5a placed them in the opposite facial orientation. Therefore, the D/E rings adopted the trans positional relationship [13, 14], and consequently 13-OH was in the α-orientation of ring D. Furthermore, the ROESY correlations between H3-26 and H-5b, between H3-26 and gem-dimethyl H3-28/H3-29, implied the a-orientation of H-9.
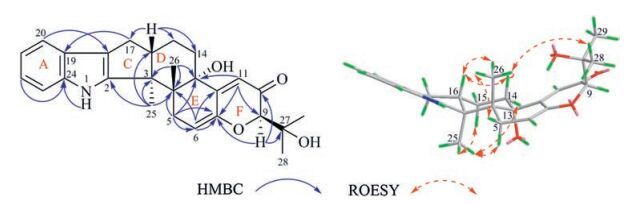
The absolute configuration of compound 1 was determined by the circular dichroism (CD) exciton chirality method [15-17]. In the CD spectrum of compound 1 (Fig. 3), a negative cotton effect of the α, β, γ, δ-unsaturated ketone (C-6/7/12/11/10) at 239 nm (Δε -21.2) and a positive cotton effect of the indole moiety at 224 nm (Δε +7.0) were observed. The negative chirality arose from the anticlockwise manner of above two chromophores in space, which was only possible under the 3S, 4R, 13S, and 16S configurations. Thus, compound 1 was determined as description.

|
Download:
|
| Fig. 3. The CD spectra of 1 (A) and 2 (B) in MeOH. | |
Compound 2 was isolated as a pale yellow powder. On the basis of positive HRESIMS ion at m/z 462.2385 [M+H]+ (calcd. for C27H32N3O4, 462.2387), its molecular formula was determined as C27H31N3O4. Analysis of its 1D NMR data established the similar indole-diketopiperazine to 10 and 11, especially when observing the 1, 2, 4-trisubstituted benzene ring, the two prenyl, the proline residue, and three amide/carbonyl alike carbons (Table 1). But since the additional olefinic proton at δH 5.72 (br s, 1H, H-10) in 2 gave HMBC correlations (Fig. 4) to C-11 and C-19, the double bond at Δ10(11) was established. More importantly, H-10 showed HMBC correlations to C-4, C-20 and the distinctive quaternary carbon C-3 at δC 61.4, and both H-20 and H2-25 had HMBC correlations with the carbonyl C-2 at δC 176.7. These key evidence indicated that the sprio[pyrrolidin-3, 3'-oxindole] ring system in 2 was similar to spirotryprostatin B [18, 19], which also explained the HMBC correlations from H-20 to C-3, C-4 and C-10. Further analyzing the HMBC correlations confirmed its planar structure.

|
Download:
|
| Fig. 4. The key HMBC and ROESY correlations of 2. | |
The relative configuration of 2 was elucidated as the same as that of spirotryprostatin B. The ROESY correlations (Fig. 4) between H-5/H-21/H-20 and H-10, indicated that the two planes of the sprioring system were arranged in an approximately vertical manner and that H-5 and H-21 are in close spatial proximity. The ROESY correlation between H-14 and H-20 showed that they were in the same facial orientation.
The CD exciton chirality method was also used to determine the absolute configuration of 2. In its CD spectrum (Fig. 3), a positive cotton effect was observed, which originated from the exciton coupling between chromophores of the α, β-unsaturated amide (C-10/11/19) at 242 nm (Δε +62.0) and the oxindole moiety at 221 nm (Δε -46.7). The clockwise manner of two chromophores in space established the absolute configuration of C-3 (3S). Combined with the relative configuration, C-14 and C-20 were therefore determined as 14S and 20S, respectively. Thus, 2 was identified as the derivative of spirotryprostatin B, and was named spirotryprostatin F.
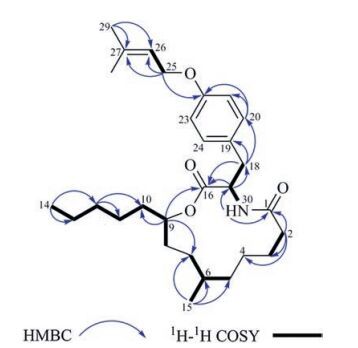
Compound 3 was obstained as colorless amorphous solid. The molecular formula of 3, C29H45NO4, was determined through the positive ion at m/z 494.3237 [M+Na]+, which indicated 8° of unsaturation. Although the molecular formula indicated that 3 was another alkaloid, its NMR data was quite different from those of other isolates. Its 1D NMR data showed signals of a 1, 4-disubstituted benzene ring, a prenyl, and two carbonyls. These above functional groups account for 7 indices of hydrogen deficiency, which meant that the remaining sixteen aliphatic carbons only occupied one degree of unsaturation. Thus 3 might be a macrolide. According to detial 2D NMR analysis, 3 was established to have the same skeleton as melearoride A, PF1163A, and PF1163B [20-22]. The HMBC correlations (Fig. 5) from H-9 to C-7, C-10 and C-16 indicated the ester bond between C-9 and C-16. The NH signal at δH 5.68 (d, J = 9.5 Hz) was confirmed by the HMBC correlations from δH 5.68 to C-1 (δC 173.0) and C-17 (δC 53.8), which also approved the amido linkage of C-1/C-17. Meanwhile, the HMBC correlations from H2-25 to C-22, C-26 and C-27, indicated an oxygenated prenyl to C-22, and the HMBC correlations from H3-15 to C-5, C-6 and C-7 indicated a methyl to C-6.

|
Download:
|
| Fig. 5. The key HMBC and 1H-1H COSY correlations of 3. | |
Same as those for the known analogues the key ROESY correlations of H-6/H-9/H-10a and of H-17/H-10b indicated that H-6 and H-9 were on the same face of the macrolide ring system, while H-17 was in the opposite direction. The same relative configurations along with the similar negative rotation (
All compounds (1-11) were evaluated for their cytotoxic activities against three human tumor cell lines (Table 2). The new compounds 2 and 3 showed moderate activities to the MDAMB-231 and HepG2 cell lines. The structure-activity relationships of 4 and its analogues (1, 5, 6, and 7) indicated that the presence of α, β-unsaturated carbonyl obviously decreased the cytotoxicities to all three cell lines. Meanwhile, indole-diketopiperazines with 11b-hydroxyl in 11 showed most potent cytotoxicities, followed by 11ahydroxyl in 8, 9, and 10 successively, which indicated that the presence of the 11β-hydroxyl in 11 showed most potent cytotoxicities, followed by 11α-hydroxyl in 8, 9, and 10 successively, which indicated that the presence of the 11β-hydroxyl was important for their cytotoxicities.
|
|
Table 2 Cytotoxicities of compounds 1–11 in three human tumor cell lines with MTT method. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3. Conclusion
Three new alkaloids, 6, 7-dehydropaxilline (1), spirotryprostatin F (2), and N-demethylmelearoride A (3), along with 8 known alkaloids were isolated from the endophytic P. brefeldianum. These metablites are biosynthetically through different pathways that combined polyketide, peptide and terpenoid biosynthesis. The indoloditerpenes and the 13-membered macrolide were reported from this species for the fist time, which enriched the metabolic diversity of Penicillium sp. In addition, some of these compounds showed moderate cytotoxicities against human tumor cell lines, which should be further investigated.
4. Experimental 4.1. General experimental proceduresOptical rotations were measured with a Jasco P-1020 polarimeter. UV spectra were acquired with a Shimadzu UV-2450 spectrophotometer. CD spectra were recorded on a Jasco J-810 spectrometer. IR spectra (KBr disks, in cm-1) were obtained on a Bruker Tensor 27 spectrometer. The NMR spectra (1H: 500 MHz, 13C: 125 MHz) were measured at 300 K with a Bruker Avance Ⅲ NMR instrument with TMS as internal standard. CDCl3 (δH = 7.26 and δC = 77.16 for CDCl3) was used as solvent. Mass spectra were obtained on an Agilent 6520 B UPLC-Q-TOF spectrometer. Column chromatography (CC) was carried out using silica gel (Qingdao Marine Chemical Co. Ltd., Qingdao, China), Sephadex LH-20 (Pharmacia, Stockholm, Sweden), RPC18 silica (40-63 mm, FuJi, Japan). The fractions obtained from column chromatography were detected by TLC with precoated silica gel GF254 (Qingdao Haiyang Chemical Co., Ltd., China) plates. Analytical HPLC was performed with an Agilent 1200 Series instrument with a DAD detector by using a shim-pack VPODS column (4.6 mm × 250 mm; Agilent Technologies, Palo Alto, CA, USA). Preparative HPLC was carried out using a Shimadzu LC-8A equipped with a Shim-pack RP-C18 column (20 mm × 200 mm, i.d., Shimadzu, Tokyo, Japan) with a flow rate of 10.0 mL/min, detected by a binary channel UV detector.
4.2. Fungal materials and fermentationThe fungal strain was isolated from the rhizome of Pinellia ternata that collected from suburb of Nanjing, Jiangsu Province, People's Republic of China, in October 2014. It was identified as Penicillium brefeldianum by the morphological method and sequence analysis of internal transcribed spacer (ITS) rDNA (GenBank accession No. KM243921.1). The strain was cultured on potato dextrose agar (PDA) at 28 ℃. After 7 days, agar plugs were cut into small pieces (about 0.3 cm × 0.3 cm × 0.3 cm) under aseptic condition, 10-15 pieces of which were inoculated to a Erlenmeyer flask (500 mL), containing 200 mL of potato dextrose liquid medium. Then the flasks were incubated on a rotary shaker at 28 ℃ and 170 rpm for 6 days to prepare the seed culture. Fermentation was carried out in 10 Erlenmeyer flasks (500 mL), each containing 80 g of rice and 120 mL distilled H2O, and cultivated at 28 ℃ for 35 days. The solid culture medium was soaked overnight and sterillizing at 121 ℃ for 20 min before using.
4.3. Extraction and isolationThe solid rice material was extracted with equivoluminal EtOAc three times at room temperature, and the organic solvent was evaporated to dryness under vacuum to afford the crude extract (13.0 g). The EtOAc extract was fractionated by silica gel CC using petroleum ether-EtOAc (v/v, 1:0, 60:1, 40:1, 20:1, 10:1, 5:1, 2:1, 1:1 and 0:1) gradient elution to give nine fractions (Fr.1-Fr.9). Fr.6 (497.7 mg) was submitted to Sephadex LH-20 CC with CH2Cl2-MeOH (1:1) as eluent, giving seven subfractions (a-g) after combination according to TLC. The subfraction Fr.6-f (104.0 mg) was further purified by preparative HPLC with MeOH-H2O (87:13), to give 4 (51.0 mg, tR = 15.6 min), 5 (5.5 mg, tR = 6.9 min), 7 (3.9 mg, tR = 9.1 min), respectively. The subfraction Fr.6-g (18.9 mg) was further purified by preparative HPLC with MeOH-H2O (78:22), to give 6 (10.9 mg, tR = 13.8 min). The subfraction Fr.6-d (199.8 mg) was further fractionated by medium pressure preparative HPLC and purified subsequently by preparative HPLC with MeOH-H2O (87:13), to give 3 (5.1 mg, tR = 18.5 min). Fr.7 (577.7 mg) was also submitted to Sephadex LH-20 CC with CH2Cl2-MeOH (1:1) as eluent, giving six subfractions (a-f) after combination according to TLC. The subfraction Fr.7-e (38.7 mg) was further purified by preparative HPLC with MeOH-H2O (67:33), to give 1 (5.7 mg, tR = 26.4 min). The subfraction Fr.7-c (281.1 mg) was further purified by medium pressure preparative HPLC with MeOH-H2O (66:34), to give 8 (10.9 mg, tR = 24.3 min) and 10 (8.6 mg, tR = 45.1 min). The subfraction Fr.7-b (105.1 mg) was further purified by preparative HPLC with MeOH-H2O (82:18), to give 9 (5.1 mg, tR = 15.4 min). Fr.8 and Fr.9 were combined to over a reversedphase C18 column with MeOH-H2O (v/v, 30:70-100:0) to obtain six subfractions (Ca-Cf). The subfraction Cc (65.0 mg) was further purified by preparative HPLC with MeOH-H2O (65:35), to give 2 (2.6 mg, tR = 17.6 min) and 11 (2.6 mg, tR = 31.5 min).
Compound 1: Yellow powder; [α]D 25 +20.0 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 204 (4.06), 229 (4.18), 277 (3.53), 321(3.38) nm; IR (KBr) vmax 3435, 2922, 1638, 1384, 1262, 1171, 1109, 622 cm-1; CD (MeOH) λmax (Δε) 208 (-17.6), 224 (+7.0), 239 (-21.2), 263 (+10.9) 327 (+29.2) and 372 (-17.3) nm; 1H and 13C NMR data, see Table 1; HR-ESIMS m/z 434.2325 [M+H]+ (calcd. for C27H32NO4, 434.2326).
Compound 2: Pale yellow powder; [α]D 25 -27.8 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 205 (4.14), 216(4.15), 271 (3.68) nm; IR (KBr) vmax 3447, 2925, 2853, 1638, 1383, 1098 cm-1; CD (MeOH) λmax (Δε) 202 (-13.1), 221(-46.7), 242 (+62.0), 271 (-18.4) and 310 (+1.4) nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 462.2385 [M+H]+ (calcd. for C27H32N3O4, 462.2387).
Compound 3: Colorless amorphous solid; [α]D 25 -55.9 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 204 (4.18), 226 (4.07), 277(3.16) nm; IR (KBr) vmax 3422, 1729, 1640, 1513, 682 cm-1; 1H and 13C NMR data, see Table 1; HR-ESIMS m/z 494.3237 [M+Na]+ (calcd. for C29H45NNaO4, 494.3241).
4.4. Bioactivity assayCytotoxic activities were evaluated against three human tumor cell lines by the MTT method [25]. Cisplatin and doxorubicin were used as the positive control medicine.
AcknowledgmentsThis research work was funded by the National Natural Science Foundation of China (No. 81503218), the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT_15R63), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Fundamental Research Funds for the Central Universities (No. 2016ZZD010).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.02.022.
| [1] | D. Hoffmeister, N.P. Keller. Natural products of filamentous fungi:enzymes, genes, and their regulation. Nat. Prod. Rep. 24(2007)393–416. DOI:10.1039/B603084J |
| [2] | J. Schümann, C. Hertweck. Molecular basis of cytochalasan biosynthesis in fungi:gene cluster analysis and evidence for the involvement of a PKS-NRPS Hybrid synthase by RNA silencing. J. Am. Chem. Soc. 129(2007)9564–9565. DOI:10.1021/ja072884t |
| [3] | A. Gallo, M. Ferrara, G. Perrone. Phylogenetic study of polyketide synthases and nonribosomal peptide synthetases involved in the biosynthesis of mycotoxins. Toxins 5(2013)717–742. DOI:10.3390/toxins5040717 |
| [4] | S. White, J. O'Callaghan, A. D.W. Dobson. Cloning and molecular characterization of Penicillium expansum genes upregulated under conditions permissive for patulin biosynthesis. FEMS Microbiol. Lett. 255(2006)17–26. DOI:10.1111/fml.2006.255.issue-1 |
| [5] | T.T. Bladt, J.C. Frisvad, P.B. Knudsen, T.O. Larsen. Anticancer and antifungal compounds from Aspergillus, Penicillium and other filamentous fungi. Molecules 18(2013)11338–11376. DOI:10.3390/molecules180911338 |
| [6] | S.T. Chen, J.F. Wang, X.P. Lin, et al., Chrysamides A-C, three dimeric nitrophenyl trans-epoxyamides produced by the deep-sea-derived fungus Penicillium chrysogenum SCSIO41001. Org. Lett. 18(2016)3650–3653. DOI:10.1021/acs.orglett.6b01699 |
| [7] | L.H. Meng, C.Y. Wang, A. Mándi, et al., Three diketopiperazine alkaloids with spirocyclic skeletons and one bisthiodiketopiperazine derivative from the mangrove-derived endophytic fungus Penicillium brocae MA-231. Org. Lett. 18(2016)5304–5307. DOI:10.1021/acs.orglett.6b02620 |
| [8] | M. Sasaki, M. Tsuda, M. Sekiguchi, Y. Mikami, J. Kobayashi, A. Perinadine. a novel tetracyclic alkaloid from marine-derived fungus Penicillium citrinum. Org. Lett. 7(2005)4261–4264. DOI:10.1021/ol051695h |
| [9] | Y.C. Zhang, C. Li, D.C. Sweson, et al., Novel antiinsectan oxalicine alkaloids from two undescribed fungicolous Penicillium spp. Org. Lett. 5(2003)773–776. DOI:10.1021/ol0340686 |
| [10] | J.W. Zhu, H. Nagasawa, F. Nagura, et al., Elucidation of strict structural requirements of brefeldin A as an inducer of differentiation and apoptosis. Bioorg. Med. Chem. 8(2000)455–463. DOI:10.1016/S0968-0896(99)00297-7 |
| [11] | J.W. Zhu, H. Hori, H. Nojiri, T. Tsukudaa, Z. Taira. Synthesis and activity of brefeldin a analogs as inducers of cancer cell differentiation and apoptosis. Bioorg. Med. Chem. Lett. 7(1997)139–144. DOI:10.1016/S0960-894X(96)00588-4 |
| [12] | T. Hosoe, K. Nozawa, S.I. Udagawa, S. Nakajima, K.I. Kawai. Structures of new indoloditerpenes possible biosynthetic precursors of the tremorgenic mycotoxins, penitrems, from Penicillium crustosum. Chem. Pharm. Bull. 38(1990)3473–3475. DOI:10.1248/cpb.38.3473 |
| [13] | T. Itabashi, T. Hosoe, D. Wakana, et al., A new indoloditerpene derivative penijanthine A, isolated from Penicillium janthinellum. J. Nat. Med. 63(2009)96–99. DOI:10.1007/s11418-008-0292-6 |
| [14] | D.M. Roll, L.R. Barbieri, R. Bigelis, et al., The lecanindoles, nonsteroidal progestins from the terrestrial fungus Verticillium lecanii 6144. J. Nat. Prod. 72(2009)1944–1948. DOI:10.1021/np9004882 |
| [15] | N. Berova, L.D. Bari, G. Pescitelli. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 36(2007)914–931. DOI:10.1039/b515476f |
| [16] | F.L. An, J. Luo, X.B. Wang, et al., Trichiconlides A and B:two novel limonoids from the fruits of Trichilia connaroides. Org. Biomol. Chem. 14(2016)1231–1235. DOI:10.1039/C5OB02300A |
| [17] | S. Lhamo, X.B. Wang, T.X. Li, et al., Three unusual indole diketopiperazine alkaloids from a terrestrial-derived endophytic fungus, Aspergillus sp. Tetrahedron Lett. 56(2015)2823–2826. DOI:10.1016/j.tetlet.2015.04.058 |
| [18] | A. Awata, T. Arai. Catalytic asymmetric exo'-selective[3+2] cycloaddition for constructing stereochemically diversified spiro[pyrrolidin-3.30-oxindole]s. Chem, Eur. J. 18(2012)8278–8282. DOI:10.1002/chem.v18.27 |
| [19] | C.B. Cui, H. Kakeya, H. Osada. Novel mammalian cell cycle inhibitors spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron 52(1996)12651–12666. DOI:10.1016/0040-4020(96)00737-5 |
| [20] | M. Okabe, T. Sugita, K. Kinoshita, K. Koyama. Macrolides from a marine-derived fungus, Penicillium meleagrinumvar. viridiflavum, showing synergistic effects with fluconazole against azole-resistant Candida albicans. J. Nat. Prod. 79(2016)1208–1212. DOI:10.1021/acs.jnatprod.6b00019 |
| [21] | H. Nose, A. Seki, T. Yaguchi, et al., PF1163A and B new antifungal antibiotics produced by Penicillium sp. I. taxonomy of producing strain. fermentation, isolation and biological activities. J. Antibiot. 53(2000)33–37. DOI:10.7164/antibiotics.53.33 |
| [22] | T. Sasaki, H. Nose, A. Hosoya, et al., PF1163A and B, new antifungal antibiotics produced by Penicillium sp. Ⅱ. physico-chemical properties and structure elucidation. J. Antibiot. 53(2000)38–44. DOI:10.7164/antibiotics.53.38 |
| [23] | F. Bouazza, B. Renoux, C. Bachmann, J.P. Gesson. Total synthesis and conformational analysis of the antifungal agent (-)-PF1163B. Org. Lett. 5(2003)4049–4052. DOI:10.1021/ol035309c |
| [24] | F. Bouazza, B. Renoux, C. Bachmann, J.P. Gesson. Total synthesis and conformational analysis of the antifungal agent (-)-PF1163B. Org. Lett. 6(2004)1069. DOI:10.1021/ol049946u |
| [25] | C. Zhang, L. Yang, X.B. Wang, et al., Calyxin Y induces hydrogen peroxidedependent autophagy and apoptosis via JNK activation in human non-small cell lung cancer NCI-H460 cells. Cancer Lett. 340(2013)51–62. DOI:10.1016/j.canlet.2013.06.021 |