 2017, Vol. 28
2017, Vol. 28 



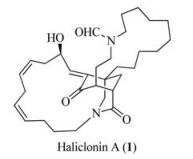
In 2009, Oh, Shin, and co-workers reported the isolation of (-)-haliclonin A (1) (Fig. 1), a tetracyclic natural product, from a marine sponge Haliclona sp. collected from Korean waters [1]. In connection with our interest in the synthesis of bioactive natural products [2], we have embarked on the synthesis of (-)-haliclonin A. In 2013, we communicated a racemic synthesis of a tricyclic core of haliclonin A (1) [3]. Lately, we have disclosed the first enantioselective total synthesis of (-)-haliclonin A (1) [4].

|
Download:
|
| Fig. 1. Structure of (-)-haliclonin A. | |
{kind=link}
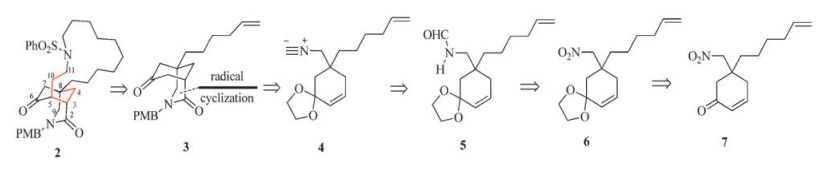
In connection with our interest in the development of synthetic methodologies for the direct transformation of amides [5], we are interested in exploring an alternative approach for the efficient construction of the tricyclic core (2) of haliclonin A. The new approach is outlined retrosynthetically in Scheme 1. Inspired by Bachi's thiol-medicated free radical cyclization of alkenyl isocyanides [6] to give γ-lactams [6c-g], bridged-bicyclic intermediate 3 was envisioned to be accessible from isonitrile 4. The latter in turn would be available from formamide 5 by dehydration [7]. Formamide 5 could be synthesized from nitro-enone 7 via its protected form, namely, acetal derivative 6.

|
Download:
|
| Scheme1. Retrosynthetic analysis of 2. | |
{kind=link}
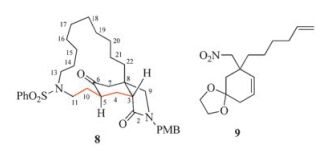
According to the retrosynthetic analysis, a synthetic route was planned and executed. However, after finishing the synthetic sequence, it appeared to us that the final product was not the desired tricyclic lactam 2. To elucidate the structure of the final synthetic product, an investigation by means of various 2D NMR techniques [8] was undertaken, which revealed its structure to be that represented by 8 (Fig. 2). This result implicated that the acetal formed from 2-cyclohexenone 7 was 9 instead of 6. These results allowed displaying a correct synthetic route. The results of this investigation are described in this report.

|
Download:
|
| Fig. 2. Structures of compound 8 and 9. | |
{kind=link}
2. Results and discussion 2.1. Structural elucidation of the final synthetic product 8 by 2D NMR techniques
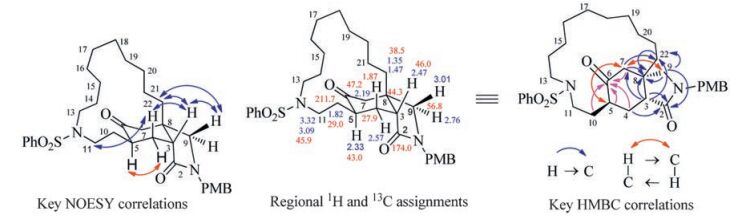
The absent of vinyl resonances in the low field zone of 4.80-6.60 ppm in the 1H NMR spectrum of the RCM product, and the appearance of three more methylene resonances in the aliphatic region of 13C NMR spectrum indicated that the expected macrocycle has been formed smoothly. The azabicyclononane core of the expected product 2 featured a CH2-CH-CH-CH2-CH2 segment (C4-C3-C5-C10-C11) and two isolated methylenes (C7, C9). The resonances appeared at δH 2.76, 3.01 were assigned to the C9 methylene according to the observed correlations with lactam carbonyl (C2) at δC 174.0 in the HMBC spectrum. Likewise, HMBC correlation between ketone carbonyl (C6) at δC 211.7 and another lone methylene resonance at δH 2.19 allowed us to identify C7. The methylene at δH 3.09, 3.32 showed only two COSY cross peaks to the methylene at δH 1.82, furthermore, the HMBC correlation was observed between δH 1.82 and δC 211.7. Therefore, the resonances at δH 3.09, 3.32 and at δH 1.82 were assigned to H11(CH2) and H10 (CH2), respectively. The observed COSY cross peaks at δH10 1.82/δH 2.33(CH); δH 2.33/δH 1.87, 2.57(CH2); δH 1.87, 2.57/δH 2.47(CH, triplet) allowed us to identify a CH-CH2-CH-CH2-CH2 segment. However, this was not in agreement with the CH2-CH-CH-CH2-CH2 segment of the expected product 2. This evidence allowed concluding that the product did not possess an azabicyclononane core, i.e., the final product was not the expected 2.
Next we proceeded to assign the structure of the unexpected product. The HMBC cross peaks at δH 2.19(H7)/δC 43.0(C5); δH 2.33 was determined by the NOESY correlations between δH3 2.47/δβ-H4 1.87; δβ-H4 1.87/δH11 3.09, 3.32; δH5 2.33/δα-H4 2.57. Thus the structure of the unexpected di-aza-tricyclic product was elucidated to be 8 (Fig. 3).

|
Download:
|
| Fig. 3. Key 1H and 13C NMR data and key correlations in the NOESY and HMBC spectra of compound 8. | |
{kind=link}
2.2. Execution of the synthetic plan
The synthesis started from the known nitro-enone 7 [3, 4] (Scheme 2). At the outset of our investigation the mild Hwu's protocol [9] that we have previously used for the synthesis of a tricyclic core of sarain A was adopted [2d]. However, when nitroenone 7 was treated with 1, 2-bis[(trimethylsilyl)oxy]ethane (BTSE) and a catalytic amount of trimethylsilyl trifluoromethanesulfonate (TMSOTf) at -20 ℃ for 12 h, the starting material remained intact. To our delight, by running the reaction at room temperature for 12 h, a dioxolanation product was obtained in 80% yield. Alternatively, we also examined the simpler classical protocol. Thus, p-toluene sulfonic acid (PTSA, 0.25 equiv.) catalyzed condensation of 7 with ethylene glycol in refluxing toluene with removal of water formed with a Dean-Stark apparatus produced the same acetal in 90% yield. Interestingly, replacing PTSA by pyridinium p-toluenesulfonate (PPTS) afforded the same major product (78% yield), along with an isomer in 10% yield. The 1H NMR and 13C NMR of the two isomers are quite similar. In the light of the report [9] that TMSOTf-mediated dioxolanation of α, β--unsaturated aldehydes and ketones yielded chemoselectively simple acetal as the major products, and the olefinic bond migrated acetals being the minor products, the major product of our reaction has been by error assigned as acetal 6. The ultimate elucidation of the structure of 8 allowed revising the major acetal to 9 (vide supra). Without having recognized this problem, the synthesis has been pursued, which is now described according to the revised result.

|
Download:
|
| Scheme2. Unexpected synthesis of 8 from a route planned for the synthesis of 2. | |
{kind=link}
The nitro group in 9 was reduced with lithium aluminum hydride [10] in diethyl ether at r.t. for 4 h to give the corresponding primary amine, which, without further purification, was heated in neat ethyl formate at reflux [11] overnight to produce formamide 10 in 85% yield over two steps. Dehydration of formamide 10 with phosphoryl chloride (POCl3) and diisopropylamine [7c] at 0 ℃ for 2 h afforded the presumed isonitrile 11, which, without further purification, was subjected to Bachi's cyclization conditions [6f, g]. However, when 11 was treated with 0.15 equiv. of azoisobutyronitrile (AIBN) and 2.0 equiv. of 2-mercaptoethanol in degassed and anhydrous toluene at 40 ℃ for 12 h, formamide 10 was recovered as the major product. To our delight, when running the reaction at 80 ℃ for 12 h, the cyclization occurred partially to give the cyclization product 12 in 45% yield, along with 40% of formamide 10. To improve yield of the cyclization, a high dilution technique was adopted. Thus, by slowly injecting of a toluene solution of 2-mercaptoethanol (2.0 equiv.) through an injecting pump to a mixture of AIBN (0.15 equiv.) and isocyanate 11 in toluene at 80 ℃, the desired product 12 was obtained in 72% yield. Further improvement consisted in replacing AIBN with 1, 1'-azobis (cyclohexanecarbonitrile) (ACCN) as a radical initiator, and slow addition of 2-mercaptoethanol (2.0 equiv.) through an injecting pump over 4 h, followed by stirring the resulting mixture at 85 ℃ for 12 h. In this manner, the desired cyclization product 12 was obtained in 80% yield.
With the bicyclic lactam 12 in hand, our next task was the introduction of an alkenyl side chain at α-position of the ketone group. For this purpose, secondary lactam 12 was deprotonated with NaH and allowed reacting with p-methoxybenzyl bromide. By quenching the reaction with 3 mol/L HCl, and stirring the resulting mixture at 50 ℃ for 6 h, keto-lactam 13 was delivered directly in 75% overall yield.
Now we were in a position to undertake a ketone a-functionalization of 13 to give diene 15. Unfortunately, attempted alkylation of 13 with functionalized alkyl iodide 14a was unsuccessful under a number of conditions including using a variety of bases (LDA, LHMDS, NHMDS, KHMDS, tBuOK), solvents (CH2Cl2, THF, toluene, DMF), reaction temperatures, as well as addition of HMPA as an additive. In all cases, only the starting ketone was recovered. The failure could be attributed to steric hindrance of the bicyclic ring system for the alkylation via a SN2 mechanism. To overcome this difficulty, an aldol condensation reaction was envisioned as an alternative. In fact, it is well known that an aldehyde is much more reactive than an alkyl halide. In this regard, in the formal synthesis of the englerins, Gao and Cook have achieved the direct aldol condensation [12] of a hydroazulenone with acetaldehyde under soft enolization conditions (TiCl4-Hünig base) to form the corresponding enone [12a]. Indeed, by successively treating ketone 13 with TiCl4 (1.2 equiv.), Hünig base (1.3 equiv.) (-78 ℃, 2 h), and aldehyde 14b (-78 ℃, 2 h, then 35 ℃, 24 h), the aldol condensation reaction proceeded smoothly to produce regioselectively the dienic α, β-enone 16 as an inseparable mixture of geometric isomers in 65% yield. The structures of these compounds were deduced from that of 8. Because the newly formed olefinic bond was to be reduced in the subsequent step, the mixture of geometric isomers was used in the next step without further separation. We next proceeded to investigate the RCM reaction [13, 14] to build the macrocycle. To avoid possible intermolecular reaction, high dilution conditions were adopted. Thus, a mixture of diene-enone 15 and Grubb's 2nd Generation catalyst (0.3 equiv.) [15] in CH2Cl2 (0.0003 mol/L) was heated at 40 ℃ for 12 h to afford the corresponding macrocyclization product (no shown) as a mixture of several geometric isomers in 60% yield. Catalytic hydrogenation (H2, 1 atm, 10% Pd/C, EtOAc, r.t., 4 h) of the mixture afforded tricyclic keto-lactam 8 in 98% yield.
3. ConclusionOn the basis of 2D NMR techniques COSY, HSQC, HMBC and NOESY, the structure of the final product of the novel synthetic route designed to access the tricyclic core of haliclonin A was determined. As a result, the product turned out to be not the desired one (2), but compound 8. Although this result is unexpected, the efficient strategy to access the novel tricyclic compound 8 is of value. In particular, it provides a novel application of the Bachi's thiol-medicated free radical cyclization of alkenyl isocyanides to build the hexahydro-1H-isoindole-1, 5 (4H)-dione ring system. It is worth mentioning that this ring system represents a drug-like chiral bicyclic scaffold [16], which has attracted much synthetic attention [17]. The result allows demonstrating the potential and power of the Bachi's cyclization reaction in building novel ring system from simple and easily available starting materials. In addition, we have shown that aldol condensation could serve as an effective alternative to an unsuccessful alkylation reaction in a congested ring system without increasing any synthetic step.
4. ExperimentalGeneral methods are described in the previous paper [3].
To a solution of enone 7 (4.74 g, 20 mmol) in toluene (130 mL) were added ethylene glycol (6.21 g, 100 mmol) and p-toluene sulfonic acid monohydrate (PTSA) (965 mg, 5.0 mmol). The flask was fitted to a Dean-Stark apparatus and the setup was purged with argon. After being refluxed for 24 h, the reaction was cooled to room temperature and diluted with diethyl ether (100 mL). The resulting solution was successively washed with a saturated aqueous solution of NaHCO3 (2 × 10 mL), water and brine. The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc/hexane = 1/25) to give compound 9 (5.06 g, yield: 90%).
When pyridinium p-toluenesulfonate (PPTS) was used in place of PTSA, compound 9 was obtained in 78% yield, along with 6 in 10% yield.
[9-(Hex-5-en-1-yl)-9-(nitromethyl)]-1, 4-dioxaspiro[4.5]dec-7-ene (9): Pale yellow oil. 1H NMR (400 MHz, CDCl3): δ 5.80-5.72 (m, 2H, CH=CH2 and CCH=CH), 5.40-5.36 (m, 1H, CCH=CH), 5.00-4.89 (m, 2H, CH=CH2), 4.70 (d, 1H, J = 10.7 Hz, NCH2), 4.43 (d, 1H, J = 10.7 Hz, NCH2), 4.08--4.02 (m, 1H, OCH2), 3.98-3.91 (m, 3H, OCH2), 2.35-2.18 (m, 2H, CH=CHCH2), 2.05-1.95 (m, 3H, CH=CHCH2 and COCH2C), 1.69 (d, 1H, J = 14.2 Hz, COCH2C), 1.52-1.23 (m, 6H, CH2); 13C NMR (125 MHz, CDCl3): δ 138.5, 129.1, 126.7, 114.5, 107.5, 81.4, 64.5, 64.1, 41.7, 37.6, 36.6, 35.5, 33.4, 29.1, 22.9; IR (film): vmax 2934, 1546, 1376, 1141, 909, 739 cm-1; MS (ESI) m/z (%): 304 (100) [M + Na]+; HRMS (ESI) calcd. for [C15H23NO4 + Na]+: 304.1519; found: 304.1525.
9-(Hex-5-en-1-yl)-9-(nitromethyl)-1, 4-dioxaspiro-[4.5]dec-6-ene (6): Pale yellow oil. 1H NMR (400 MHz, CDCl3): δ 5.89-5.83 (m, 1H, COCH=CH), 5.78-5.74 (m, 1H, CH=CH2), 5.63-5.58 (m, 1H, COCH=CH), 5.01-4.89 (m, 2H, CH=CH2), 4.53 (d, 1H, J = 11.8 Hz, NCH2), 4.45 (d, 1H, J = 11.8 Hz, NCH2), 3.97-3.86 (m, 4H, OCH2), 2.27-2.18 (m, 1H, CH=CHCH2), 2.07-1.94 (m, 3H, CH=CHCH2 and CH=CHCH2), 1.93 (d, 1H, J = 14.0 Hz, COCH2C), 1.82 (d, J = 14.0 Hz, 1H, COCH2C), 1.52-1.28 (m, 6H, CH2); 13C NMR (125 MHz, CDCl3): δ = 138.5, 128.9, 127.1, 114.5, 104.5, 80.0, 64.5, 64.3, 40.6, 38.2, 37.2, 33.4, 33.1, 29.0, 22.3; IR (film): vmax = 2934, 1546, 1376, 1141, 909, 739 cm-1; MS (ESI) m/z (%): 304 (100) [M + Na]+; HRMS (ESI) calcd. for [C15H23NO4 + Na]+: 304.1519; found: 304.1525.
To a suspension of LiAlH4 (2.28 g, 60.0 mmol) in Et2O (100 mL) was slowly added a solution of compound 9 (4.22 g, 15.0 mmol) in Et2O (10 mL) at 0 ℃. After being stirred at room temperature for 6 h, the resulting mixture was cooled to 0 ℃ and Na2SO4·10H2O (30.0 g) was added in 10 portions. After 15 min of stirring, the mixture was filtered through a pad of Celite and the residue was washed with CH2Cl2 for eight times. Removal of the solvent under reduced pressure. Without further purification, to the residue (3.61 g, 14.4 mmol) was added ethyl formate (50 mL) and the mixture was refluxed for 12 h. The reaction mixture was then cooled to room temperature and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc/hexane = 1/1) to give formamide 10 (3.54 g, yield: 85%) as a pale yellow sticky oil.
(S)-N-[(7-(Hex-5-en-1-yl)-1, 4-dioxaspiro[4.5]dec-8-en-7-yl) methyl]formamide (10): 1H NMR (400 MHz, CDCl3, data of the major rotamer read from the spectrum of the mixture): δ 8.12 (s, 1H, CHO), 6.57 (br s, 1H, NH), 5.80-5.64 (m, 2H, CH=CH and CH=CH2), 5.36-5.29 (m, 1H, CH=CH), 4.99-4.86 (m, 2H, CH=CH2), 4.00-3.86 (m, 4H, OCH2), 3.41 (dd, 1H, J = 13.4, 7.0 Hz, NCH2), 3.33 (dd, 1H, J = 13.4, 4.4 Hz, NCH2), 2.24-2.16 (m, 2H, CH=CHCH2), 2.04-1.96 (m, 2H, CH2CH=CH2), 1.80 (d, 1H, J = 14.3 Hz, OCCH2C), 1.63 (d, 1H, J = 14.3 Hz, OCCH2C), 1.40-1.17 (m, 6H, CH2); 13C NMR (125 MHz, CDCl3): δ 161.3, 138.6, 132.0, 124.9, 114.3, 108.1, 64.5, 64.0, 46.8, 41.3, 39.9, 39.5, 34.8, 33.5, 29.2, 22.9; IR (film): vmax 3329, 2930, 1666, 1536, 1385, 1362, 1080, 914, 802, 728 cm-1; MS (ESI) m/z (%): 302 (100) [M + Na]+; HRMS (ESI) calcd. for [C16H25NO3 + Na]+: 302.1727; found: 302.1724.
To a solution of formamide 10 (1.39 g, 5 mmol) in CH2Cl2 (50 mL) was added iPr2NH (3.49 mL, 25 mmol) and the mixture was cooled to 0 ℃. To the mixture was added dropwise POCl3 (0.93 mL, 10 mmol) and the resulting mixture was stirred at 0 ℃ for 2 h. The reaction was quenched with a saturated aqueous solution of NaHCO3 (10 mL) and the resulting mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was used in the next step without further purification. To a solution of the residue and 1, 1'-azobis(cyclohexanecarbonitrile) (ACCN) (244 mg, 1 mmol) in dry toluene (250 mL) at 85 ℃ was slowly added a solution of 2-mercaptoethanol in dry toluene (20 mL) with a syringe pump over 4 h and the resulting mixture was stirred at the same temperature for another 12 h. After being cooled to room temperature, the reaction was quenched with a solution of saturated NaHCO3 (100 mL) before being extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc/hexane = 1/1) to give bicyclic compound 12 (1.11 g, yield: 80%) as a pale yellow sticky oil.
(3a'S, 7a'S)-3a'-(Hex-5-en-1-yl)hexahydrospiro[[1, 3]dioxolane-2, 5'-isoindol]-1'(6'H)-one (12): 1H NMR (400 MHz, CDCl3): δ 6.85 (br s, 1H, NH), 5.77 (ddt, 1H, J = 17.0, 10.3, 6.8 Hz, CH=CH2), 5.01-4.91 (m, 2H, CH=CH2), 3.92-3.84 (m, 4H, OCH2), 3.04 (d, 1H, J = 9.5 Hz, NCH2), 2.93 (d, 1H, J = 9.5 Hz, NCH2), 2.14-2.09 (m, 1H, NCOCH), 2.09-2.01 (m, 4H, CH2CO2 and CH2CH=CH2), 1.90 (ddd, 1H, J = 13.1, 4.0, 3.0 Hz, NCOCHCH2), 1.82-1.75 (m, 1H, NCOCHCH2), 1.73 (d, 1H, J = 14.0 Hz, CCH2C), 1.56 (d, 1H, J = 14.0 Hz, CCH2C), 1.50-1.28 (m, 5H, CH2), 1.17-1.03 (m, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ 179.0, 138.6, 114.5, 108.5, 64.5, 63.7, 51.5, 46.2, 43.5, 37.7, 36.7, 33.4, 31.3, 29.2, 23.6, 19.4; IR (film): vmax = 3230, 2928, 1701, 1440, 1365, 1254, 1095, 950, 822 cm1; MS (ESI) m/z (%): 302 (100) [M + Na]+; HRMS (ESI) calcd for [C16H25NO3 + Na]+: 302.1727; found: 302.1727.
To a suspension of NaH (60% dispersion in mineral oil, 500 mg, 12.5 mmol) in THF (45 mL) was slowly added a solution of secondary lactam 12 (1.39 g, 5.0 mmol) in THF (5 mL) at 0 ℃. After being stirred at room temperature for 0.5 h, the resulting mixture was cooled to 0 ℃ and p-methoxybenzyl bromide (1.09 mL, 7.5 mmol) was added dropwise. After the reaction was finished as indicated by TLC monitoring, the mixture was cooled to 0 ℃ and quenched with 3 mol/L HCl (20 mL), which was then heated to 50 ℃ and stirred for another 6 h. After being cooled to room temperature, the reaction was slowly quenched with a solution of saturated NaHCO3 (50 mL) and then extracted with EtOAc (3 × 30 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc/hexane = 1/1) to give bicyclic compound 13 (1.33 g, yield: 75%) as a pale yellow sticky oil.
(3aS, 7aR)-3a-(Hex-5-en-1-yl)-2-(4-methoxybenzyl)hexa-hy-dro-1H-isoindole-1, 5(6H)-dione (13): 1H NMR (500 MHz, CDCl3): δ 7.16-7.12 (m, 2H, Ar-H), 6.87-6.83 (m, 2H, Ar-H), 5.73 (ddt, 1H, J = 16.8, 10.3, 6.7 Hz, CH=CH2), 4.98-4.90 (m, 2H, CH=CH2), 4.46 (d, 1H, J = 14.4 Hz, PhCH2), 4.34 (d, 1H, J = 14.4 Hz, PhCH2), 3.78 (s, 3H, CH3), 3.01 (d, 1H, J = 10.2 Hz, CCH2N), 2.75 (d, 1H, J = 10.2 Hz, CCH2N), 2.47-2.40 (m, 2H, NCOCH and NCOCHCH2), 2.29 (d, 1H, J = 14.4 Hz, COCH2C), 2.27-2.21 (m, 2H, COCH2), 2.18 (d, 1H, J = 14.4 Hz, COCH2C), 2.05-1.95 (m, 3H, NCOCHCH2 and CH2CH=CH2), 1.52-1.09 (m, 6H, CH2), 13C NMR (125 MHz, CDCl3): δ 210.8, 174.1, 159.2, 138.3, 129.6 (2C), 128.0, 114.8, 114.1 (2C), 56.0, 55.2, 47.0, 46.8, 46.1, 42.1, 39.3, 37.1, 33.4, 29.0, 23.2, 22.4; IR (film): vmax = 2931, 1668, 1611, 1513, 1445, 1247, 1176, 1034, 910, 810 cm-1; MS (ESI) m/z (%): 378 (100) [M + Na]+; HRMS (ESI) calcd. for [C22H29NO3 + Na]+: 378.2040; found: 378.2039.
To a cooled (-78 ℃) solution of ketone 13 (71.1 mg, 0.20 mmol) in CH2Cl2 (2 mL) was added dropwise TiCl4 (1.0 mol/L in CH2Cl2, 0.22 mL, 0.22 mmol) under N2. After 15 min, iPr2NEt (45 μL, 0.24 mmol) was added dropwise, and the resulting deep red solution was stirred at -78 ℃ under N2 for 2 h. After dropwise addition of a solution of aldehyde 14b [4] (112 mg, 0.40 mmol) in CH2Cl2 (0.5 mL), the resulting mixture was stirred at -78 ℃ for 2 h and then warm slowly to 35 ℃. The resulting mixture was stirred at that temperature for another 24 h. After being cooled to 0 ℃, the reaction was quenched by addition of a solution of saturated aqueous NH4Cl, and the resulting mixture was extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4 filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc/hexane = 1/2) to give enone 16 (76.6 mg, yield: 65%) as a colorless sticky oil.
N-(Hex-5-en-1-yl)-N-(2-((3aS, 7aS)-7a-(hex-5-en-1-yl)-2-(4-methoxybenzyl)-3, 6-dioxohexahydro-1H-isoindol-5(6H)-ylidene)ethyl)-benzenesulfonamide (16): 1H NMR (500 MHz, CDCl3, data of the major isomer read from the spectrum of the mixture of geometric isomers): δ 7.82-7.78 (m, 2H, Ar-H), 7.59-7.48 (m, 3H, ArH), 7.04-7.01 (m, 2H, Ar-H), 6.85-6.81 (m, 2H, Ar-H), 6.55 (dd, 1H, J = 6.7, 6.2 Hz, CH=CCO), 5.77-5.67 (m, 2H, CH=CH2), 4.98-4.89 (m, 4H, CH=CH2), 4.31 (d, 1H, J = 14.6 Hz, PhCH2), 4.27 (d, 1H, J = 14.6 Hz, PhCH2), 4.03 (dd, 1H, J = 17.3, 6.7 Hz, NCH2CH), 3.87 (dd, 1H, J = 17.3, 6.2 Hz, NCH2CH), 3.78 (s, 3H, CH3), 3.10 (t, 2H, J = 7.5 Hz, NCH2), 3.02 (d, 1H, J = 10.4 Hz, NCH2C), 2.98 (dd, 1H, J = 17.7, 6.8 Hz, NCOCHCH2), 2.82 (d, 1H, J = 10.4 Hz, NCH2C), 2.62-2.58 (m, 1H, NCOCH), 2.49 (d, 1H, J = 14.4 Hz, COCH2), 2.29 (d, 1H, J = 14.4 Hz, COCH2), 2.03-1.96 (m, 5H, CH2CH=CH2 and NCOCHCH2), 1.57-1.29 (m, 8H, CH2), 1.24-1.14 (m, 2H, CH2); 13C NMR (125 MHz, CDCl3): δ 197.6, 173.9, 159.2, 139.5, 138.21, 138.17, 135.0, 133.9, 132.6, 129.34 (2C), 129.26 (2C), 129.1, 127.6, 127.1 (2C), 114.8, 114.1 (2C), 56.6, 55.2, 48.5, 47.4, 46.7, 45.9, 45.6, 41.8, 37.8, 33.3, 33.1, 28.9, 27.3, 25.7, 25.1, 23.0; IR (film): vmax 2930, 1686, 1513, 1446, 1341, 1247, 1160, 1091, 1030, 910, 752, 694, 574 cm-1; MS (ESI) m/z (%): 641 (100) [M + Na]+; HRMS (ESI) calcd. for [C36H46N2O5S +Na]+: 641.3020; found: 641.3028.
A 500 mL flask charged with CH2Cl2 (250 mL) and diene 16 (46.4 mg, 0.075 mmol) was purged with argon. The solution was heated to 40 ℃, and a solution of Grubbs second generation catalyst (10 mg, 0.011 mmol) in CH2Cl2 (2 mL) was added in one portion. After being stirred for 12 h under the same temperature, another solution of catalyst (10.2 mg, 0.011 mmol) in CH2Cl2 (2 mL) was added in one portion. After being stirred for another 12 h, the solution was allowed cooling to room temperature over 2 h, then the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc/ CH2Cl2 = 1/3) to give the RCM product (35 mg, yield: 60%) as an inseparable mixture of geometric isomers. 1H NMR (500 MHz, CDCl3, data of the major isomer read from the spectrum of the mixture): δ 7.82-7.76 (m, 2H, Ar-H), 7.62-7.50 (m, 3H, Ar-H), 7.13-7.09 (m, 2H, Ar-H), 6.85-6.81 (m, 2H, Ar-H), 6.80-6.76 (m, 1H, CH=CCO), 5.35-5.08 (m, 2H, CH=CH), 4.42 (d, 1H, J = 14.4 Hz, PhCH2), 4.32 (d, 1H, J = 14.4 Hz, PhCH2), 3.77 (s, 3H, CH3), 3.57-3.37 (m, 2H, NCH2CH), 3.12-2.99 (m, 3H, NCH2 and CCH2N), 2.73-2.62 (m, 1H, NCHCH2), 2.49-2.33 (m, 4H, COCH2 and COCH and NCOCH), 2.09-1.80 (m, 4H, CH2CH=CH), 1.63-1.18 (m, 10H, CH2), 1.15-1.09 (m, 2H, CH2) 13C NMR (125 MHz, CDCl3): δ 197.2, 173.9, 159.2, 135.0, 133.7, 132.7, 130.6, 130.3, 129.4 (2C), 129.2 (2C), 128.0, 127.02, 127.0 (2C), 114.1 (2C), 59.4, 55.2, 51.5, 48.4, 47.7, 45.9, 45.2, 41.7, 37.9, 31.9, 31.2, 29.1, 27.7, 27.3, 25.6, 22.1; IR (film): vmax 2928, 1737, 1688, 1445, 1246, 1162, 1092, 1034, 972, 737, 577 cm-1; MS (ESI) m/z (%): 613 (100) [M + Na]+; HRMS (ESI) calcd. for [C34H42N2O5S +Na]+: 613.2707; found: 613.2718.
Palladium on carbon (25.0 mg, 10% Degüssa type) was added to a degassed solution of the RCM product (24.2 mg, 0.041 mmol) in EtOAc (4 mL). The flack was purged with H2 and the suspension was stirred under H2 (1 atm) at room temperature for 4 h. The reaction mixture was filtered through a plug of SiO2 topped with Celite (eluent: EtOAc). The filtrate was concentration under reduced pressure to afford compound 8 (24.5 mg, yield: 98%) as a white solid. Mp 88-90 ℃ (EtOAc); 1H NMR (500 MHz, CDCl3): δ 7.82-7.76 (m, 2H, Ar-H), 7.56-7.46 (m, 3H, Ar-H), 7.16-7.02 (m, 2H, Ar-H), 6.86-6.82 (m, 2H, Ar-H), 4.49 (d, 1H, J = 14.4 Hz, PhCH2), 4.31 (d, 1H, J = 14.4 Hz, PhCH2), 3.78 (s, 3H, CH3), 3.39-3.25 (m, 2H, NCH2CH2C and NCH2CH2CH2), 3.21-3.14 (m, 1H, NCH2CH2CH2), 3.10-3.03 (m, 1H, NCH2CH2C), 3.01 (d, 1H, J = 10.0 Hz, CCH2N), 2.76 (d, 1H, J = 10.0 Hz, CCH2N), 2.58-2.51 (m, 1H, NCOCHCH2), 2.45 (dd, 1H, J = 5.3, 4.8 Hz, NCOCH), 2.35-2.27 (m, 1H, COCH), 2.20 (d, 1H, J = 14.2 Hz, COCH2), 1.97 (d, 1H, J = 14.2 Hz, COCH2), 1.89-1.83 (m, 1H, NCOCHCH2), 1.83-1.75 (m, 2H, NCH2CH2), 1.56-1.42 (m, 2H, NCH2CH2), 1.39-1.12 (m, 16H, CH2); 13C NMR (125 MHz, CDCl3): δ 211.6, 174.0, 159.3, 140.3, 132.2, 129.6 (2C), 129.0 (2C), 128.1, 127.0 (2C), 114.2 (2C), 56.8, 55.2, 48.7, 47.2, 46.2, 46.0, 45.9, 44.3, 43.0, 38.5, 29.0, 28.2, 28.1, 27.9, 27.73, 27.69, 27.6, 26.4, 25.0, 23.0; IR (film): vmax 2930, 1691, 1607, 1513, 1446, 1335, 1246, 1158, 1088, 1030, 744, 686, 582 cm-1; MS (ESI) m/zm/z (%): 617 (100) [M + Na]+; HRMS (ESI) calcd. for [C34H46N2O5S +Na]+: 617.3020; found: 617.3024.
AcknowledgementThe authors are grateful to the National Natural Science Foundation of China (No. 21472153), the National Basic Research Program (973 Program) of China (No. 2010CB833200), the SKL of Xiamen University (No. 201509), and to the Program for Changjiang Scholars and Innovative Research Team in University of Ministry of Education, China, for financial support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.04.016.
| [1] | K.J. Jang, G.W. Kang, J. Jeon, et al., Haliclonin A:a new macrocyclic diamide from the sponge Haliclona sp. Org. Lett. 11(2009)1713–1716. DOI:10.1021/ol900282m |
| [2] |
(a) P.Q. Huang, Z.Y. Mao, H. Geng, Enantioselective total synthesis and structural revision of (-)-isochaetominine, Chin. J. Org. Chem. 36(2016) 315-324; (b) J.L. Ye, Y. Liu, Z.P. Yang, P.Q. Huang, Asymmetric total synthesis of (+)-Nacetyl norloline, Chem. Commun. 52(2016) 561-563; (c) Z.Y. Mao, H. Geng, T.T. Zhang, et al., Stereodivergent and enantioselective total syntheses of isochaetominines A-C and four pairs of isochaetominine C enantiomers:a six-step approach, Org. Chem. Front. 3(2016) 24-37; (d) R.F. Yang, P.Q. Huang, Studies towards an enantioselective total synthesis of sarain A:a concise asymmetric construction of the diazatricyclic core, Chem.-Eur. J. 16(2010) 10319-10322. |
| [3] |
(a) S.P. Luo, L.D. Guo, L.H. Gao, S. Li, P.Q. Huang, Toward the total synthesis of haliclonin A:construction of a tricyclic substructure, Chem.-Eur. J. 19(2013) 87-91; (b) S.P. Luo, Ph.D. Dissertation, Studies on the Total Syntheses of Haliclonin A, Chaetominine, Asperlicin E and the Syntheses of Sulindac Analogs, Xiamen University, 2012. |
| [4] | L.D. Guo, X.Z. Huang, S.P. Luo, et al., Organocatalytic. asymmetric total synthesis of (-)-haliclonin a. Angew. Chem. Int. Ed. 55(2016)4064–4068. DOI:10.1002/anie.201512005 |
| [5] |
(a) P.Q. Huang, W. Ou, F. Han, Chemoselective reductive alkynylation of tertiary amides by Ir and Cu(I) bis-metal sequential catalysis, Chem. Commun. 52(2016) 11967-11970; (b) Q.W. Lang, X.N. Hu, P.Q. Huang, Tf2O-TMDS combination for the direct reductive transformation of secondary amides to aldimines, aldehydes, and/or amines, Sci. China Chem. 59(2016) 1638-1644; (c) P.Q. Huang, Y.H. Huang, H. Geng, J.L. Ye, Metal-free C—H alkyliminylation and acylation of alkenes with secondary amides, Sci. Rep. 6(2016) 28801; (d) P.Q. Huang, Y.H. Huang, K.J. Xiao, Metal-free intermolecular coupling of arenes with secondary amides:chemoselective synthesis of aromatic ketimines and ketones, and N-deacylation of secondary amides, J. Org. Chem. 81(2016) 9020-9027; (e) A.E. Wang, Z. Chang, Y.P. Liu, P.Q. Huang, N-Deacylation of secondary amides by alkylation with organocerium reagents, Chin. Chem. Lett. 24(2015) 1055-1058; (f) J.F. Zheng, Z.Q. Xie, X.J. Chen, P.Q. Huang, Direct transformation of amides:reductive cycloaddition of secondary amides with Danishefsky diene, Acta Chim. Sinica 73(2015) 705-715; (g) H. Geng, P.Q. Huang, Versatile and chemoselective transformation of aliphatic and aromatic secondary amides to nitriles, Tetrahedron 71(2015) 3795-3801. |
| [6] |
(a) F. Dénès, M. Pichowicz, G. Povie, P. Renaud, Thiyl radicals in organic synthesis, Chem. Rev. 114(2014) 2587-2693; (b) R. Wang, H. Jiang, Y.Z. Cheng, et al., Somophilic isocyanide insertion:synthesis of 6-arylated and 6-trifluoromethylated phenanthridines, Synthesis 46(2014) 2711-2726; (c) M.D. Bachi, A. Melman, Enantioselective total synthesis of (-)-α-kainic acid, Pure & Appl, Chem. 70(1998) 259-262; (d) M.D. Bachi, A. Melman, Enantioselective total synthesis of (-)-α-kainic acid through free radical cyclization of an alkenyl monothioformimide, J. Org. Chem. 62(1997) 1896-1898; (e) M.D. Bachi, N. Bar-Ner, A. Melman, Stereoselective synthesis of ((±)-α-kainic acid using free radical key reactions, J. Org. Chem. 61(1996) 7116-7124; (f) M.D. Bachi, A. Melman, Stereocontrolled 5-exo-trig cyclization of imidoyl radicals in the synthesis of substituted (alkylthio)pyrrolines, pyroglutamates, and thiopyroglutamates, J. Org. Chem. 60(1995) 6242-6244; (g) M.D. Bachi, A. Balanov, N. Bar-Ner, Thiol mediated free radical cyclization of alkenyl and alkynyl isocyanides, J. Org. Chem. 59(1994) 7752-7758. |
| [7] |
(a) X. Wang, Q.G. Wang, Q.L. Luo, Synthesis of isonitriles from N-substituted formamides using triphenylphosphine and iodine, Synthesis 47(2015) 49-54; (b) J.E. Baldwin, I.A. O'Neil, Trifluoromethanesulfonic anhydride, a superior reagent for the conversion of formamides to isonitriles, Synlett (1990) 603-604; (c) R. Obrecht, R. Hermann, I. Ugi, Isocyanide synthesis with phosphoryl chloride and diisopropylamine, Synthesis (1985) 400-401. |
| [8] |
(a) I. Noda, Recent developments in two-dimensional (2D) correlation spectroscopy, Chin. Chem. Lett. 26(2015) 167-172; (b) C. Long, W.C. Luo, H.Y. Zhou, et al., Isolation of toxic compounds from wild Phaeocystis globosa, Chin. Chem. Lett. 27(2016) 247-250. |
| [9] |
(a) J.R. Hwu, L.C. Leu, J.A. Robl, D.A. Anderson, J.M. Wetzel, General scope of 1, 3-dioxolanation of α, β-unsaturated aldehydes with 1, 2-bis(trimethylsilyloxy) ethane and trimethylsilyl trifluoromethanesulfonate, J. Org. Chem. 52(1987) 188-191; (b) J.R. Hwu, J.M. Wetzel, The trimethylsilyl cationic species as a bulky proton. Application to chemoselective dioxolanation, J. Org. Chem. 50(1985) 3946-3948. |
| [10] | J. Hubner, J. Liebscher, M. Patzel. Patzel M.. Optically active nitroalkenes-synthesis. addition reactions and transformation into amino acids. Tetrahedron 58(2002)10485–10500. DOI:10.1016/S0040-4020(02)01406-0 |
| [11] | C.S. Swindell, B.P. Patel. Stereoselective construction of the taxinine AB system through a novel tandem aldol-Payne rearrangement annulations. J. Org. Chem. 55(1990)3–5. DOI:10.1021/jo00288a002 |
| [12] |
(a) P. Gao, S.P. Cook, A reductive-Heck approach to the hydroazulene ring system:a formal synthesis of the englerins, Org. Lett. 14(2012) 3340-3343; (b) R. Mahrwald, Diastereoselection in Lewis-acid-mediated aldol additions, Chem. Rev. 99(1999) 1095-1120. |
| [13] | A. Deiters, S.F. Martin. Synthesis of oxygen-and nitrogen-containing heterocycles by ring-closing metathesis. Chem. Rev. 104(2004)2199–2238. DOI:10.1021/cr0200872 |
| [14] | M. Becker, H.P. Chua, R. Downham, et al., Total synthesis of (-)-sarain a. J. Am. Chem. Soc. 129(2007)11987–12002. DOI:10.1021/ja074300t |
| [15] | A.K. Chatterjee, T.L. Choi, D.P. Sanders, R.H. Grubbs. A general model for selectivity in olefin cross metathesis. J. Am. Chem. Soc. 128(2003)11360–11370. |
| [16] | G. Mariaule, S.D. Cesco, F. Airaghi, et al., 3-oxo-Hexahydro-1H-isoindole-4-carboxylic acid as a drug chiral bicyclic scaffold:structure-based design and preparation of conformationally constrained covalent and noncovalent prolyl oligopeptidase inhibitors. J. Med. Chem. 59(2016)4221–4234. DOI:10.1021/acs.jmedchem.5b01296 |
| [17] |
(a) B. Richey, K.M. Mason, M.S. Meyers, S.B. Luesse, Rapid access to conformationally-constrained oxatricycles via Ugi-Smiles couplings, Tetrahedron Lett. 57(2016) 492-494; (b) A. Tomberg, S.D. Cesco, M. Huot, N. Moitessier, Solvent effect in diastereoselective intramolecular Diels-Alder reactions, Tetrahedron Lett. 56(2015) 6852-6856; (c) X.J. Fang, R. Jackstell, M. Beller, Sequential hydroformylation/Diels-Alder processes:one-pot synthesis of polysubstituted cyclohexenes, cyclohexadienes, and phthalates from alkynes, Chem.-Eur. J. 20(2014) 7939-7942; (d) C.X. Da, W.G. Shou, Y.G. Wang, Microwave-promoted Beller synthesis of Nsubstituted 2, 3, 3a, 4, 7, 7a-hexahydroisoindole-1, 3-diones, Chin. J. Chem. 24(2006) 689-694. |