 2017, Vol. 28
2017, Vol. 28 



Aminopeptidases represent a class of zinc-containing metal loenzymes responsible for the cleavage of amino acids from the N-terminus of various peptides or proteins [1–3]. The relationship between aminopeptidases and cancer was first reported in the late 1950s [4, 5], which attracted the attention of many scientists in the following dozen years. The first aminopeptidase inhibitor bestatin was discovered in 1976 (Fig. 1) [6], and it was subsequently used as an effective anti-cancer agent. Notwithstanding many other aminopeptidase inhibitors in clinical trials, bestatin is so far the only clinically approved one in Japan used for anti-tumor treatment [7]. Recently, it is suggested that aminopeptidases play significant roles in regulating the supply of cellular free amino acids to maintain the survival and proliferation of cancer cells [8]. Especially, cancer cells are dependent on some specific amino acids and the deprivation of these amino acids may provide a simple, effective and non-genotoxic strategy for anticancer therapy [9]. Thus, the development of novel aminopeptidase inhibitors for the treatment of cancer is still of tremendous potential.

|
Download:
|
| Figure 1. Bestatin and representative compounds with 1, 2, 3-triazin-4-one and 4-thiazolidinone scaffolds | |
Five key aminopeptidases—aminopeptidase N (APN), leucine aminopeptidase (LAP), puromycin-sensitive aminopeptidase (PuSA), leukotriene A4 hydrolase (LTA4H) and endoplasmic reticulum aminopeptidase 1/2 (ERAP1/2) are now deemed to have close correlation with a wide range of cancers [8]. LTA4H, a monomeric zinc-containing soluble protein, acts as a dual-function enzyme with both epoxide hydrolase and aminopeptidase activi ties [10, 11]. In the 5-lipoxygenase pathway of arachidonic acid metabolism, LTA4H catalyzes the conversion of unstable epoxide Leukotriene A4 to diol Leukotriene B4 [12], which is associated with a variety of inflammatory diseases [13–15]. As an epoxide hydrolase, LTA4H has long been a crucial target in anti inflammatory drug discovery. Besides, LTA4H exhibits a significant sequence homology to some other zinc peptidases such as aminopeptidase M, aminopeptidase B, and thermolysin [3, 16]. On the basis of its zinc binding motif—HEXXHX18E [17, 18], LTA4H was proved to possess the peptide-cleaving activity as a member of the M1 family of zinc aminopeptidases [19]. To the best of our knowledge, there is currently rare effective aminopeptidase inhibitors specifically acting on LTA4H. Therefore, the search for novel LTA4H aminopeptidase inhibitors to obtain potential anticancer agents is valuable.
1, 2, 3-Triazin-4-one represents a privileged structure in medici nal chemistry, which serves as a unique building block for the construction of many compounds with anti-cancer activity (Fig. 1, 1–4). Compounds 2–4 were reported to have matrix metalloproteinase (MMP) inhibitory activity [20–22]. Among them, compound 3 showed good activity in vivo and had a potential for further development [22]. It is well-known that MMPs are also zinc-containing metalloenzymes and share a similar catalytic mechanism with LTA4H. So, we consider that 1, 2, 3-triazin-4-one may be used as a possible pharmacophore in our design of novel LTA4H aminopeptidase inhibitors. Furthermore, 4-thiazolidinone is another important motif found in a number of bioactive compounds [23]. For example, compound 5 could selectively kill both non-small cell lung cancer cell line H460 and its paclitaxel resistant variant H460taxR [24], and compound 6 showed promising cytotoxicity against renal cancer cell lines (UO-31, TK-10, and ACHN) [25]. More importantly, due to LTA4H has an L-shaped substrate binding pocket [26], we speculate that a flexible carbon chain of appropriate length may be necessary to make the entire molecule fit the binding pocket well. Thereupon, the above two fragments were combined by a flexible carbon chain to design our target molecule (Fig. 2). Thirty-three novel 1, 2, 3-benzotriazin-4-one derivatives with 4-thiazolidinone moieties were herein synthesized and their bioactivity evaluation against LTA4H aminopeptidase was performed. The influence of linker length and substituent to inhibitory activity was also investigated.

|
Download:
|
| Figure 2. Design strategy of novel target compound Ⅳ. | |
2. Results and discussion 2.1. Synthesis
The synthetic procedure of the target compounds Ⅳ-1–32 is illustrated in Scheme 1. Using various substituted anthranilamides Ⅰ-1–11 as starting materials, substituted 1, 2, 3-benzotriazin-4-ones Ⅱ-1–11 could be easily prepared in one pot via diazotization, nucleophilic addition and cyclization [27] in yields of above 80%. Subsequently, in the presence of potassium carbonate, Ⅱ-1–11 reacted with dibromoalkane (2–6 carbons) in acetone to obtain 3-bromoalkane-1, 2, 3-benzotriazin-4-ones [28] Ⅲ-1–33 in yields of 46%–62%. As an alkylation agent, dibromoalkane exhibits two reaction sites, but we obtained the N-alkylation of compound Ⅲ-1-33, N-3 alkylated ones were the main products. Clark et al. predicted 1, 2, 3-benzotriazin-4(3H)-one to be the most stable of its three tautomers [27]. To confirm the structures of N-3 alkylated products, 2D NMR (HMBC) was performed. From the HMBC spectrum (500 MHz) of 3-(3-bromopropyl)-7-chlorobenzo[d] [1, 2, 3]triazin-4(3H)-one, it can be seen that the methylene (H-5) is strongly related to the carbonyl group (C-4) of benzotriazinone but not related to the benzene ring (The corresponding spectra are deposited in Supporting information). Because dibromoalkane can readily react with NH at the 3-position of 1, 2, 3-benzotriazin-4-one to afford a N3, N3'-(alkane-1, 3-diyl)bis(benzo[d][1, 2, 3]triazin-4 (3H)-one), to minimize the incidence of this side reaction, different amounts of dibromoalkane were tried. It was found that when dibromoalkane was of excess up to 5 equiv., the generation of the N3, N3'-(alkane-1, 3-diyl)bis(benzo[d][1, 2, 3]triazin-4(3H)-one)

|
Download:
|
| Scheme 1. General synthetic route for target compound Ⅳ. | |
could be well inhibited. Then the target compounds Ⅳ-1–33 were synthesized via the reaction of 3-bromoalkane-1, 2, 3-benzotriazin-4-ones Ⅲ-1–33 with 2-cyanoimino-4-oxothiazolidine potassium salt. The addition of potassium iodide was profitable to the above nucleophilic substitution owing to better leaving ability of iodide ion than bromide ion. The chemical structures of Ⅳ-1–33 were characterized by 1H NMR, 13C NMR, 19F NMR and HRMS (see Supporting information).
2.2. Biological activityThe LTA4H aminopeptidase inhibitory activities of compounds Ⅳ-1–33 were evaluated in vitro at a con-centration of 10 μmol/L by the method reported in the literature [29]. Bestatin was used as an assay positive control. The assay results are summarized in Table 1. It was found that all compounds exhibited certain inhibitory activities against LTA4H aminopeptidase. Many compounds showed > 50% inhibition rates and their IC50 values were given.
|
|
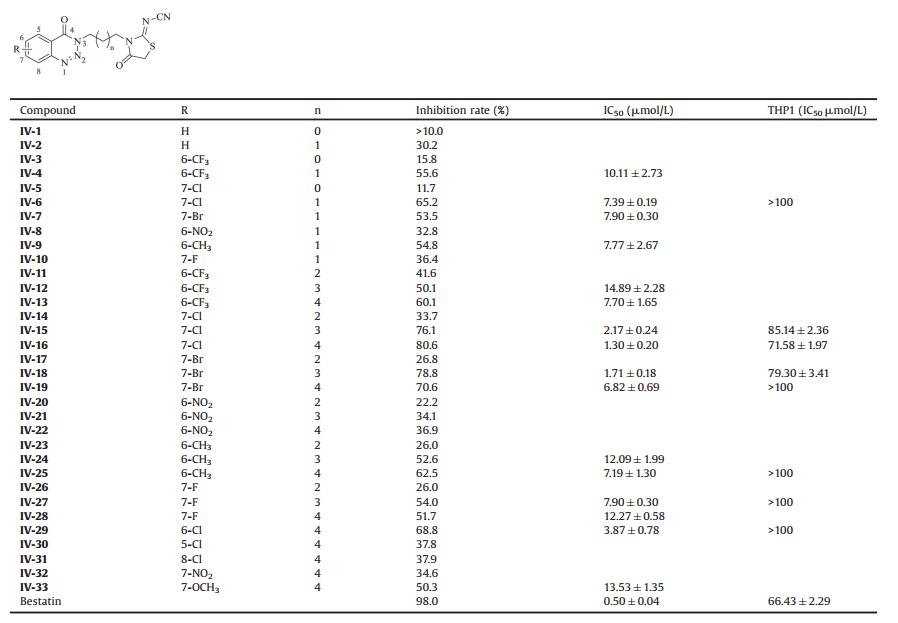
Table 1 Inhibitory activities of Ⅳ-1–33 against LTA4H aminopeptidase at 10 μmol/L. |
{kind=link}
{kind=link}
{kind=link}
As shown in Table 1, some compounds with a 2-carbon chain showed only < 20% inhibitory activities (e.g., Ⅳ-1, Ⅳ-3 and Ⅳ-5), which were much lower than those of their corresponding compounds with a 3-carbon chain (e.g., Ⅳ-1 vs. Ⅳ-2, Ⅳ-3 vs. Ⅳ-4, Ⅳ-5 vs. Ⅳ-6). It seemed that a shorter linker such as a 2-carbon chain was not beneficial to inhibitory activity. When the linker was fixed as a 3-carbon chain, it was found that the introduction of electron-withdrawing groups (CF3, NO2), electron-donating groups (CH3) and halogen (F, Cl, Br) into the benzotriazinone ring was profitable to inhibitory activity. For example, compounds Ⅳ-4 and Ⅳ-6–9 were more active than Ⅳ-2 with inhibition rates of above 30.2%. Among them, Ⅳ-6 (7-Cl) was the most potent with an IC50 of 7.39 ± 0.19 μmol/L. Considering the L-shaped pocket of LTA4H, the influence of linker length on inhibitory activity was investigated. When the length of linker was changed from 2 to 6 carbons, it was found that the activities did not increase following a certain regularity (e.g., Ⅳ-3, Ⅳ-4, Ⅳ-11–13, Ⅳ-5, Ⅳ-6, Ⅳ-14–16). However, when the length of linker was changed from 4 to 6 carbons, the majority of compounds with different substituents showed the increasing inhibitory potencies with the increasing of linker length except compounds with 7-Br (e.g., Ⅳ-11–13, Ⅳ-14– 16, Ⅳ-20–22, Ⅳ-23–25, Ⅳ-26–28). For compounds with the same substituents at the same positions on the benzotriazinone ring, a 6-carbon chain length accompanied higher activities than other chain lengths except compounds Ⅳ-19 and Ⅳ-28. For example, compounds Ⅳ-13, Ⅳ-16 and Ⅳ-25 with a 6-carbon chain had > 50% inhibitory activities with the IC50 values of 7.70 ± 1.65, 1.30 ± 0.20, and 7.19 ± 1.30 μmol/L, respectively.
Furthermore, the influence of substituted position on inhibitory activity was also investigated. When the linker length was increased from three carbons to a certain extent, Cl or Br located at 7-position on the benzotriazinone ring always seemed to provide higher potency. Notably, compound Ⅳ-16 (7-Cl) with a 6-carbon chain displayed 80.6% inhibitory activity with an IC50 of 1.30 ± 0.20 μmol/L, a little higher than Ⅳ-18 (IC50 = 1.71 ±0.18 μmol/L). It suggested that Cl group at 7-position might lead to higher activity. To verify this, the linker was fixed as a 6-carbon chain, and compounds with electron-withdrawing NO2, and electron-donating OCH3 at 7-postion were synthesized. As expected, compound Ⅳ-16 (7-Cl) was still the most potent. In addition, compounds with Cl at other three positions (5-, 6-, and 8-position) were also synthesized to examine the relationship between substituted position and inhibitory activity. The data showed that the inhibition rates of compounds with 5-and 8-Cl (Ⅳ-30, Ⅳ-31) were lower than 50%, and the IC50 of the compound with 6-Cl (Ⅳ-29) reached to 3.87 ± 0.78 mM, still lower than that of the compound with 7-Cl (IC50 = 1.30 ± 0.20 μmol/L). The com pound with 7-Cl exhibited the highest inhibitory activity among four compounds with a 6-carbon chain and differently positioned Cl (e.g., Ⅳ-16, Ⅳ-29–31).
The compounds which LTA4H aminopeptidase inhibitory IC50 better than 10 μmol/L were selected to test the proliferation inhibitory activities in THP1 human AML cell line. MTS assay showed the best three compounds in LTA4H aminopeptidase inhibition also had the better cell proliferation inhibitory activities, compound Ⅳ-16 and Ⅳ-18 were the potent compounds with 80.6% and 78.8% inhibitory activity (IC50 = 1.30 ± 0.20 μmol/L, 1.71 ±0.18 μmol/L, respectively), which both showed similar proliferation inhibitory activity as bestatin in THP1 human AML cell line.
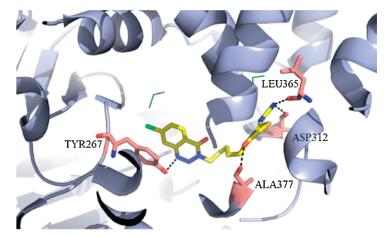
Docking study was used to explore the binding mode between the LTA4H and compound Ⅳ-16. The result of docking analysis showed that Ⅳ-16 was well docked in the active grooves of this enzyme (Fig. 3). The compound interactions with the residues in the active pocket of LTA4H mainly depending on hydrogen bonds. For example, compound Ⅳ-16 could form hydrogen bonds with the polar residues Tyr267, Ala377, Leu365 and Asp312 around the active site, which indicated that the 1, 2, 3-triazin-4-one and 4-thiazolidinone scaffolds are necessary to the inhibitory properties of compounds. And the linker of 6-carbon chain also contribute to compound Ⅳ-16 more flexible and close into the active pocket. These results are consistent with our original design.

|
Download:
|
| Figure 3. Binding poses for compound Ⅳ-16 in the binding site of LTA4H. | |
{kind=link}
From the results above, it can be concluded that 7-Cl and a longer linker length were indeed profitable to the inhibitory activity, and compound Ⅳ-16 was promising for further structural modification. Besides, a 6-carbon chain length possibly made the entire ligand molecule competitively bind the L-shaped pocket of LTA4H well and finally inhibited the aminopeptidase activity of LTA4H. Further study is now in progress.
3. ConclusionIn conclusion, a series of novel 1, 2, 3-benzotriazin-4-one derivatives were designed, synthesized and characterized. Their LTA4H aminopeptidase inhibitory activities were evaluated in vitro at the concentration of 10 μmol/L. Many compounds exhibited moderate to good inhibitory activities. In particular, compound Ⅳ-16 was the most potent with 80.6% inhibitory activity (IC50 = 1.30 ± 0.20 μmol/L), which showed similar proliferation inhibitory activity as bestatin in THP1 human AML cell line. Our results indicated that 1, 2, 3-benzotriazin-4-one scaffold was promising to study further as a lead structure for the development of LTA4H aminopeptidase inhibitors.
4. Experimental 4.1. Chemicals and instrumentsAll reagents and solvents were of AR grade orpurifiedby standard methods before use. All melting points (mp) were determined on a Büchi melting point B540 apparatus (Büchi Labortechnik AG, Flawil, Switzerland) and are uncorrected. 1H NMR, 13C NMR and 19F NMR spectra were recorded in DMSO-d6 on a Bruker AM-400 spectrome ter at ambient temperature (1H 400 MHz; 13C 100 MHz; 19F 376 MHz). Chemical shifts are reported in ppm values with tetramethylsilane (TMS) as the internal standard. Coupling con stants (J) are reported in hertz. High-resolution mass spectra (HR MS) were recorded under electro-spray ionization condition on a WatersMicromass LC-TOF spectrometer. Reactions weremonitored by thin-layer chromatography (TLC) performed on silica plates (silica gel 60 F254) using UV-light.
4.2. General synthetic procedure for target compoundsTo a mixture of 3-bromoalkyl-1, 2, 3-benzotriazin-4-one (Ⅲ, 2 mmol), and potassium iodide (0.332 g, 2 mmol) in DMF (15 mL) was added 2-cyanoimino-4-oxothiazolidine potassium salt (0.358 g, 2 mmol), and the mixture was stirred at 100 ℃. After the reaction was completed (monitored by TLC), the solvent was removed under reduced pressure to afford a dark brown oil, which was washed with water (50 mL) and extracted with CH2Cl2 (40 mL × 2). The organic layer was dried over anhydrous Na2SO4, concentrated and purified by flash chromatography on silica gel, eluting with petroleum ether (60–90 ℃)/EtOAc to afford the target compound Ⅳ. The general procedures for the synthesis of intermediates Ⅱ, Ⅲ and the spectra data of compounds Ⅳ-1–33 are listed in Supporting information.
N-(3-(4-(7-Bromo-4-oxobenzo[d][1, 2, 3]triazin-3(4H)-yl)bu tyl)-4-oxothiazolidin-2-ylidene)cyanamide (Ⅳ-16) (7-Br, 4C): yield, 10%; mp 168.5–168.8 ℃; 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.16 (d, 1H, J = 8.4 Hz), 8.09 (d, 1H, J = 8.4 Hz), 4.38 (t, 2H, J = 6.4 Hz), 4.30 (s, 2H), 3.64 (t, 2H, J = 6.4 Hz), 1.89–1.75 (m, 2H), 1.69–1.56 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 179.7, 173.1, 154.9, 145.0, 136.3, 130.7, 129.2, 127.3, 118.9, 114.2, 49.2, 42.8, 36.0, 25.8, 24.0. HRMS (ESI) calcd. for C15H12N6O2S79Br (M-H)-, 418.9926, found, 418.9929; calcd. for C15H12N6O2S81Br (M-H)-, 420.9905, found, 420.9900.
4.3. LTA4H aminopeptidase assayThe LTA4H aminopeptidase inhibitor activity assay wasmeasured by monitoring the hydrolysis of Ala-p-NA as previously described [30]. Briefly, indicated compounds were incubated in reaction buffer (50 mmol/L Tris, 100 mmol/L NaCl, pH 7.5) plus LTA4H protein (500 ng)for10 minin96-wellplatesat roomtemperature.Inthenext step, Ala-p-NA was added to initiate the reaction and recorded the the formation of p-NA in the absorption at 405 nm keeping about 10 min using Perkin Elmer Multimode Plate Reader (Evision). The IC50 values were determined from the results of at least three independent tests and calculated by the GraphPad5.
4.4. MTS assayThe antiproliferative activity of indicated compounds was measured by MTS assay as previously described [31]. The THP1 human AML cell line (5.0 ×103) were seeded into 96-well plates in 200 mL RPMI-1640 medium supplemented with 10% FBS and penicillin/streptomycin. Subsequently, different concentra tions of indicated compounds were added and incubated for another 48 h at 37 ℃ in a 5% CO2-containing atmosphere. After that, 20 mL of MTS Solution was added and incubated for 4 h, the optical density was measured at 490 nm using Perkin Elmer Multimode Plate Reader. The experiment was carried out at least three times.
4.5. Molecular dockingTo better understand the structural basis for the inhibitory activities of the inhibitors against LTA4H, we select the most potency inhibitor (Ⅳ-16) to study the binding model with LTA4H enzyme by molecular docking. The crystal structure of 4L2L was downloaded from the Protein Data Bank, then all the bound ligands and water molecules of 4L2L were removed from the complex crystal structure. Subsequently, Hydrogen atoms and charges were added using the 'AutoDockTools' and the indicated compound was located into the active pockets of LTA4H. The docking was conducted according to the instruction of AutoDock Vina described previously [32], then to select the best pose according to the scoring function. The result of docking was showed by PyMol.
AcknowledgmentsThis work was financially supported by the Na tional Natural Science Foundation of China (No. 21272071), and the Fundamental Research Funds for the Central Universities. The authors also appreciate the partial support from Shanghai Foundation of Science and Technology (No. 15431902100).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.12.014.
| [1] | A. Taylor. Aminopeptidases:structure and function. FASEB J. 7 (1993) 290–298. |
| [2] | D. Krige, L.A. Needham, L.J. Bawden, et al., CHR-2797:an antiproliferative aminopeptidase inhibitor that leads to amino acid deprivation in human leukemic cells. Cancer Res. 68 (2008) 6669–6679. DOI:10.1158/0008-5472.CAN-07-6627 |
| [3] | X.L. Jiang, L. Zhou, Y.R. Wu, et al., Modulating the substrate specificity of LTA4H aminopeptidase by using chemical compounds and small-molecule-guided mutagenesis. ChemBioChem 11 (2010) 1120–1128. DOI:10.1002/cbic.v11:8 |
| [4] | A.M. Rutenburg, J.A. Goldbarg, E.P. Pineda. Leucine aminopeptidase activityobservations in patients with cancer of the pancreas and other diseases. N. Engl. J. Med. 259 (1958) 469–472. DOI:10.1056/NEJM195809042591003 |
| [5] | R. Willighagen, H. Planteydt. Aminopeptidase activity in cancer cells. Nature 183 (1959) 263–264. DOI:10.1038/183263a0 |
| [6] | H. Umezawa, T. Aoyagi, H. Suda, M. Hamada, T. Takeuchi. Bestatin, an inhibitor of aminopeptidase b, produced by actinomycetes. J. Antibiot. 29 (1976) 97–99. DOI:10.7164/antibiotics.29.97 |
| [7] | O.A. Scornik, V. Botbol. Bestatin as an experimental tool in mammals. Curr. Drug Metab. 2 (2001) 67–85. DOI:10.2174/1389200013338748 |
| [8] | S.M. Hitzerd, S.E. Verbrugge, G. Ossenkoppele, G. Jansen, G.J. Peters. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids 46 (2014) 793–808. DOI:10.1007/s00726-013-1648-0 |
| [9] | L. Scott, J. Lamb, S. Smith, D.N. Wheatley. Single amino acid (arginine) deprivation:rapid and selective death of cultured transformed and malignant cells. Br. J. Cancer 83 (2000) 800–810. DOI:10.1054/bjoc.2000.1353 |
| [10] | B. Çalışkan, E. Banoglu. Overview of recent drug discovery approaches for new generation leukotriene A4 hydrolase inhibitors. Expert Opin. Drug Discov. 8 (2013) 49–63. DOI:10.1517/17460441.2013.735228 |
| [11] | C.D. Funk. Prostaglandins and leukotrienes:advances in eicosanoid biology. Science 294 (2001) 1871–1875. DOI:10.1126/science.294.5548.1871 |
| [12] | J.Z. Haeggstr. Structure, function, and regulation of leukotriene A4 hydrolase. Am. J. Respir. Crit. Care Med. 161 (2000) S25–S31. DOI:10.1164/ajrccm.161.supplement_1.ltta-6 |
| [13] | V.M. Tanis, G.M. Bacani, J.M. Blevitt, et al., Azabenzthiazole inhibitors of leukotriene A4 hydrolase. Bioorg. Med. Chem. Lett. 22 (2012) 7504–7511. DOI:10.1016/j.bmcl.2012.10.036 |
| [14] | N.L. Rao, P.J. Dunford, X.H. Xue, et al., Anti-inflammatory activity of a potent, selective leukotriene A4 hydrolase inhibitor in comparison with the 5-lipoxygenase inhibitor zileuton. J. Pharmacol. Exp. Ther. 321 (2007) 1154–1160. DOI:10.1124/jpet.106.115436 |
| [15] | A.M. Tager, A.D. Luster. BLT1 and BLT2:the leukotriene B4 receptors. Prostaglandins Leukot. Essent. Fatty Acids 69 (2003) 123–134. DOI:10.1016/S0952-3278(03)00073-5 |
| [16] | J.Z. Haeggström. Leukotriene A4 hydrolase/aminopeptidase, the gatekeeper of chemotactic leukotriene B4 biosynthesis. J. Biol. Chem. 279 (2004) 50639–50642. DOI:10.1074/jbc.R400027200 |
| [17] | B. Malfroy, H. Kado-Fong, C. Gros, et al., Molecular cloning and amino acid sequence of rat kidney aminopeptidase M:a member of a super family of zincmetallohydrolases. Biochem. Biophys. Res. Commun. 161 (1989) 236–241. DOI:10.1016/0006-291X(89)91586-6 |
| [18] | B.L. Vallee, D.S. Auld. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry 29 (1990) 5647–5659. DOI:10.1021/bi00476a001 |
| [19] | J.Z. Haeggström, C.D. Funk. Lipoxygenase and leukotriene pathways:biochemistry, biology, and roles in disease. Chem. Rev. 111 (2011) 5866–5898. DOI:10.1021/cr200246d |
| [20] | L. Garuti, M. Roberti, T. Rossi, M. Castelli, M. Malagoli. Synthesis and antiproliferative activity of 3-substituted lH-indole[3, 2-d]-1, 2, 3-triazin-4(3H)-ones. Eur. J. Med. Chem. 33 (1998) 43–46. DOI:10.1016/S0223-5234(99)80074-9 |
| [21] | A.M. Chollet, T.L. Diguarher, N. Kucharczyk, et al., Solid-phase synthesis of α-substituted 3-bisarylthio N-hydroxy propionamides as specific MMP inhibitors. Bioorg. Med. Chem. 10 (2002) 531–544. DOI:10.1016/S0968-0896(01)00311-X |
| [22] | T. Le Diguarher, A.M. Chollet, M. Bertrand, et al., Stereospecific synthesis of 5-substituted 2-bisarylthiocyclopentane carboxylic acids as specific matrix metalloproteinase inhibitors. J. Med. Chem. 46 (2003) 3840–3852. DOI:10.1021/jm0307638 |
| [23] | A.K. Jain, A. Vaidya, V. Ravichandran, S.K. Kashaw, R.K. Agrawal. Recent developments and biological activities of thiazolidinone derivatives:a review. Bioorg. Med. Chem. 20 (2012) 3378–3395. DOI:10.1016/j.bmc.2012.03.069 |
| [24] | H.Y. Zhou, S.H. Wu, S.M. Zhai, et al., Synthesis cytoselective toxicity, structure-activity relationships, and pharmacophore of thiazolidinone derivatives targeting drug-resistant lung cancer cells. J. Med. Chem. 51 (2008) 1242–1251. DOI:10.1021/jm7012024 |
| [25] | N. Karal, N. Karalı, A. Gürsoy. Synthesis and primary cytotoxicity evaluation of new 5-bromo-3-substituted-hydrazono-1H-2-indolinones. Arch. Pharm. 8 (2002) 374–380. |
| [26] | M.M.G.M. Thunnissen, P. Nordlund, J.Z. Haeggström. Crystal structure of human leukotriene A4 hydrolase, a bifunctional enzyme in inflammation. Nat. Struct. Mol. Biol. 8 (2001) 131–135. DOI:10.1038/84117 |
| [27] | A.S. Clark, B. Deans, M.F.G. Stevens, et al., Antitumor imidazotetrazines. 32.1 synthesis of novel imidazotetrazinones and related bicyclic heterocycles to probe the mode of action of the antitumor drug temozolomide. J. Med. Chem. 38 (1995) 1493–1504. DOI:10.1021/jm00009a010 |
| [28] | P. K. S. Sarma, S. Sharma, S. Dharmarajan, et al. , Adrenergic receptor antagonists, WO 2006117760 A1. |
| [29] | X.L. Jiang, L. Zhou, D.G. Wei, et al., Activation and inhibition of leukotriene A4 hydrolase aminopeptidase activity by diphenyl ether and derivatives. Bioorg. Med. Chem. Lett. 18 (2008) 6549–6552. DOI:10.1016/j.bmcl.2008.10.044 |
| [30] | Q. Xiao, N.N. Dong, X. Yao, et al., Bufexamac ameliorates LPS-induced acute lung injury in mice by targeting LTA4H. H, Sci. Rep. 6 (2016) 25298. DOI:10.1038/srep25298 |
| [31] | H.L. Pan, Q. Hu, J.Y. Wang, et al., Myricetin is a novel inhibitor of human inosine 50-monophosphate dehydrogenase with anti-leukemia activity. Biochem. Biophys. Res. Commun. 477 (2016) 915–922. DOI:10.1016/j.bbrc.2016.06.158 |
| [32] | O. Trott, A.J. Olson. AutoDock Vina:improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31 (2010) 455–461. |