 2017, Vol. 28
2017, Vol. 28 



b School of Biomedical Sciences, Huaqiao University, Quanzhou 362021, China
Lung cancer is the leading cause of death from cancer worldwide. Patients suffer a poor clinical outcome with five-year survival of less than 15% [1]. Non-small-cell lung cancer (NSCLC) represents a major type which accounts for approximately 80–85% of all lung cancers [2, 3].
The blockade of epidermal growth factor (EGF) signaling pathway has been identified as a promising approach for intervention of NSCLC, resulting in several therapeutic agents [4]. Gefitinib, a selective inhibitor targeting EGF receptor, has yielded great clinical benefits in patients with NSCLC, especially those harboring the EGFR-activating mutations such as delE746_A750 and L858R [5]. However, a secondary mutation T790M at the "gatekeeper" position of EGFR leads to drug resistance in much cases after 10–14 months of gefitinib administration [6, 7]. In order to overcome the resistance mechanism correlated with T790M mutation, a mount of irreversible inhibitors such as rociletinib and osimertinib have been developed, in which a acrylamide functionality was devised to undergo a Michael addition reaction with Cys797 [8–10].
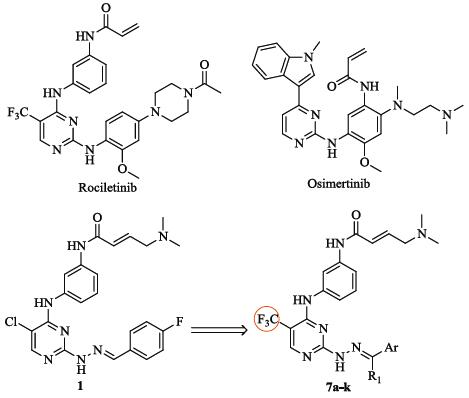
In our previous work, we reported a series of hydrazone moietybearing aminopyrimidines as selective inhibitors of EGFR T790M mutant, from which we identified a potent compound 1 (Fig. 1) as the precusor [11]. As a continuous study, we initiated a further optimization program on this promising series in order to discover compounds with more potent activity. Biological evaluations led to the identification of compounds 7f and 7k which showed significant and selective activity in inhibition of gefitinib-resistant H1975 cancer cells.

|
Download:
|
| Figure 1. Structure of rociletinib, osimertinib, and the new compounds. | |
2. Results and discussion 2.1. Chemistry
The synthesis of the target compounds 7a–k is described in Scheme 1. The intermediate 4 was synthesized via a sequential coupling of 2,4-dichloro-5-(trifluoromethyl)pyrimidine (2) with tert-butyl (3-aminophenyl)carbamate and hydrazine hydrate using a modified condition as previously reported [11]. The condensation of intermediate 4 with an appropriate aromatic aldehyde or ketone in an addition–elimination sequence generated intermediates 5a–k [12, 13], which underwent a convenient deprotection reaction to yield amines 6a–k [14]. Subsequently, amidation of 6 with commercial (E)-4-(dimethylamino)but-2-enoic acid using HATU as a dehydrant furnished the desired compounds.

|
Download:
|
| Scheme 1. Reagents and conditions: (a)DIPEA, n-butyl alcohol, 25℃, 3h; (b)NH2NH2·H2O, pyridine, ethanol, 25℃, 2 h; (c)appropriate aromatic aldehyde or ketone, ethanol, reflux, 5–7 h; (d)TFA, DCM, 25℃, 2–5 h; (e)(E)-4-(dimethylamino)but-2-enoic acid, HATU, DIPEA, DCM, 25℃, 3–10 h. | |
2.2. Design of new compounds
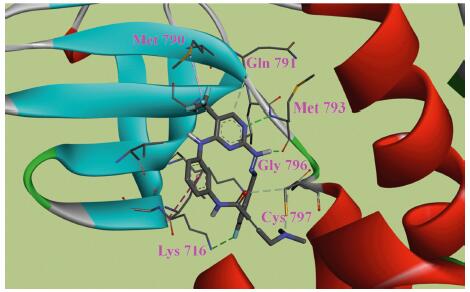
At beginning, compound 1 was docked in the active site of EGFR T790M (PDB: 3IKA) [15] to investigate the probable interactions with kinase using Discovery Studio 3.1. The results reflected that it formed anticipated hydrogen bonds with Met793 in hinge region. However, the functional acrylamide group adopted a reversed conformation, which prevented it from interacting well with Cys797. Encouraged by these observations, we initiated a program to develop an additional series of compounds with more suitable conformation to form desirable covalent bond with Cys797. In binding models of both 1 and WZ4002, a hydrophobic interaction was observed between Cl atom with Met790. We speculated that the replacement of the Cl atom with a more hydrophobic CF3 group might increase the interaction and more importantly, it could enforce the compound adopting a significantly changed conformation as compared with compound 1. To rationalize this hypothesis, a molecular docking was conducted using compound 7b. Fig. 2 showed the putative binding model with EGFR T790M, which demonstrated that apparent hydrogen-bond was formed between pyrimidine with Met793. Moreover, compound 7b adopted a very favorable conformation close to Cys797. As a consequence, an additional series of hydrazone moiety-bearing aminopyrimidines were synthesized and evaluated in both enzymatic and cellular levels with the aim to identify compounds with more potent activity.

|
Download:
|
| Figure 2. Molecular docking of compound 7b with EGFR T790M. Hydrogen bond interactions were indicated with dashed lines in green. | |
2.3. Inhibitory activity against EGFR kinase
The newly synthesized compounds were screened in a double mutant EGFR kinase, namely T790M/L858R, as well as in wild type (WT) EGFR. The enzymatic assay was carried out using a wellestablished mobility shift assay, and the results were shown in Table 1. The biological data clearly demonstrated that all the target compounds suppressed EGFR T790M/L858R in micromolar range (IC50, 0.75–5.39 µmol/L), and was more potent than gefitinib (positive control; IC50, 11.38 µmol/L). All the target compounds were not active at 10 µmol/L against wild type EGFR, suggesting a favorable kinase selectivity. Compound 7b exhibited significant activity against EGFR T790M/L858R, with IC50 value of 1.05 µmol/L, but no improvement was observed as compared with the precusor 1 (IC50, 1.13 µmol/L). Further modification on terminal benzene ring indicated that electron-donating groups in this region were better tolerated. Replacement of the fluorine atom with a methyl group led to compound 7d (IC50, 0.88 µmol/L), which was slightly more potent than 7b. Moreover, significantly increased potency could be observed when 7c (Ar=2,4-(F)2-Ph; IC50, 5.39 µmol/L) was compared with 7e (Ar=2,4-(CH3O)2-Ph; IC50, 0.77µmol/L). On the other hand, a substituent at para-position was beneficial for high activity, as shifting the fluorine atom of compound 7b to ortho-position resulted in a 4.8-fold decrease in activity (7a; IC50, 5.1 µmol/L). This effect could be explained by the putative binding model as described in Fig. 2, a hydrogen-bond was formed between fluorine atom with Lys716. The introduction of a methyl group in the hydrazone moiety (R1) was not crucial for inhibition of EGFR T790M/L858R, which indicated that the structure–activity relationship (SAR) in this region was limited. Interestingly, compound 7k containing a pyridine moiety instead of the benzene ring maintained favorable potency (IC50, 3.32 µmol/L) as well as selectivity, which highlighted the potential for further optimization.
|
|
Table 1 In vitro inhibitory profile ofcompounds 7a–k. |

{kind=link}
{kind=link}
{kind=link}
2.4. Inhibitory activity against H1975 and A549 NSCLC cells
Compounds 7a–k were screened in two type of NSCLC cancer cells including A549 (WT EGFR and K-Ras mutation) and gefitinibresistant H1975 (EGFR T790M/L858R). Notably, the attractive inhibitory activity against EGFR T790M/L858R translated well into efficacy in inhibition of H1975 cancer cells (IC50; 0.2–1.06 µmol/L), except for compound 7b, and was much more potent than that of gefitinib (IC50; 7.92 µmol/L). Also, the potency of the compounds was more pronounced against H1975 cells than against A549 cells, which correlated well with their kinase selectivity versus WT EGFR. Compound 7f demonstrated potent activity in inhibition of H1975 cancer cells, with IC50 of 0.45 µmol/L. Taken the enzymatic profile together, compound 7f was considered a promising lead in this chemical series. Also noteworthy is that compound 7k displayed the best activity in inhibition of H1975 cells (IC50; 0.2 µmol/L), while was almost inactive against A549 cells at up to 100 µmol/L. Although it possessed only moderate inhibition of EGFR T790M/L858R (IC50; 3.32 µmol/L), detailed optimization could be done to promote the potency even further.
3. ConclusionIn this paper, we provided a valuable design strategy that aimed to improve the antitumor potency of compound 1, which was identified previously as a selective inhibitor of EGFR T790M. The newly synthesized hydrazone moiety-bearing aminopyrimidines maintained favorable kinase inhibitory activity and selectivity as compared to 1. More importantly, while these compounds effectively suppressed proliferation of gefitinib-resistant H1975 cells, they showed weak effects on A549 cells, indicating a favorable selectivity. The increasement in hydrophobicity accompanied with the introduction of the CF3 group might partially account for the improved selectivity in cellular level. Compounds 7f and 7k potently and selectively inhibited proliferation of H1975 cells, and were considered promising lead compounds for further investigation.
4. Experimental 4.1. ChemistryReagents and solvents were obtained from commercial sources and used without further purification. All the reactions were monitored by TLC using silica gel GF/UV 254. Flash chromatography was performed using silica gel (300–400 mesh). The purity of the synthesized compounds was measured by high performance liquid chromatography (HPLC, Agilent, USA), and was confirmed to be higher than 95%. Melting points were determined on a Büchi Melting Point B-540 apparatus (Büchi Labortechnik, Flawil, Switzerland). The 1H and 13C NMR spectra were recorded on Bruker AV-400 spectrometer at 400 MHz and 100 MHz, respectively, with TMS as an internal standard. The low resolution of ESIMS was recorded on an Agilent 1100 LC–MS spectrometer, and high resolution mass spectrometry was performed on a Biosystems QSTAR Elite ESI-LC–MS/MS spectrometer.
4.2. Biological evaluation 4.2.1. In vitro kinase assayThe experiments were carried out by a well-established mobility shift assay, and EGFR kinases (EGFR T790M/L858R and WT EGFR) were purchased from Invitrogen. The kinase base buffer consists of 50 mmol/L HEPES (pH 7.5), 0.0015% Brij35 and 2 mmol/L DTT, while the stop buffer contained a mixture of 100 mmol/L HEPES (pH 7.5), 0.015% Brij-35, 0.2% coating reagent and 50 mmol/L EDTA. Initially, the tested compounds were diluted to 50-fold of the final desired highest concentration in reaction by 100% DMSO. Subsequently, 30 µL of the solution was transferred to 60 mL of 100% DMSO in the next well and so forth for a total of 5 concentrations. No compound and no enzyme controls were prepared by adding 100 µL DMSO to two empty wells in the same 96-well plate. Then, 10 µL of compound was transferred to a new 96-well plate, which was marked as the intermediate plate. Additional 90 µL of kinase buffer was added to each well of intermediate plate. The mixture in intermediate plate was shaked for 10 min. The assay plate was prepared after transferring 5 µL of each well from the 96-well intermediate plate to a 384-well plate in duplicates. The prepared enzyme solution (appropriate EGFR in kinase base buffer) was added to the assay plate, which was then incubated at room temperature for 10 min, followed by the addition 10 µL of prepared peptide solution (FAM-labeled peptide and ATP in kinase base buffer). The mixture was incubated at 28 ℃ for another 1 h, then 25 µL of stop buffer was added. The conversion data was copied from Caliper program, and the values were converted to inhibition values.
Percent inhibition=(max-conversion)/(max-min)×100
4.2.2. MTT cytotoxicity assayThe cancer cell lines were cultured in minimum essential medium (MEM) supplement with 10% fetal bovine serum (FBS). Approximate 4×103 cells, suspended in MEM medium, were plated onto each well of a 96-well plate and incubated in 5% CO2 at 37℃ for 24 h. The tested compounds were added to the culture medium and the cell cultures were continued for 72 h. Fresh MTT was added to each well at a terminal concentration of 5 µg/mL, and incubated with cells at 37℃ for 4 h. The formazan crystals were dissolved in 100 µL DMSO each well, and the absorbency at 492 nm (for absorbance of MTT formazan) and 630 nm (for the reference wavelength) was measured with an ELISA reader. All of the compounds were tested three times in each of the cell lines. The results expressed as IC50 (inhibitory concentration 50%) were the averages of at least three determinations and calculated by using the Bacus Laboratories Incorporated Slide Scanner (Bliss) software.
AcknowledgmentThis work was supported by grants from Science Foundation of Shenyang Pharmaceutical University (No. QNJJ2014502).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.11.030.
| [1] | R. Siegel, D. Naishadham, A. Jemal. Cancer statistics, CA Cancer J. Clin. 63 (2013) 11–30. |
| [2] | P. Praveen, H. Hülsmann, H. Sultmann, R. Kuner, H. Frohlich. Cross-talk between AMPK and EGFR dependent signaling in non-small cell lung cancer. Sci. Rep. 6 (2016) 27514. DOI:10.1038/srep27514 |
| [3] | R. Chan, P. Sethi, A. Jyoti, R. McGarry, M. Upreti. Investigating the radioresistant properties of lung cancer stem cells in the context of the tumor microenvironment. Radiat. Res. 185 (2016) 169–181. DOI:10.1667/RR14285.1 |
| [4] | K.D. Carey, A.J. Garton, M.S. Romero, et al., Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 66 (2006) 8163–8171. DOI:10.1158/0008-5472.CAN-06-0453 |
| [5] | A.J. Barker, K.H. Gibson, W. Grundy, et al., Studies leading to the identification of ZD1839(iressaTM):an orally active selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 11 (2001) 1911–1914. DOI:10.1016/S0960-894X(01)00344-4 |
| [6] | S. Kobayashi, T.J. Boggon, T. Dayaram, et al., EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352 (2005) 786–792. DOI:10.1056/NEJMoa044238 |
| [7] | C.H. Yun, K.E. Mengwasser, A.V. Toms, et al., The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 2070–2075. DOI:10.1073/pnas.0709662105 |
| [8] | A.O. Walter, R.T.T. Sjin, H.J. Haringsma, et al., Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 3 (2013) 1404–1415. DOI:10.1158/2159-8290.CD-13-0314 |
| [9] | M.R.V. Finlay, M. Anderton, S. Ashton, et al., Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 57 (2014) 8249–8267. DOI:10.1021/jm500973a |
| [10] | J.C. Chuang, A.A. Salahudeen, H.A. Wakelee. Rociletinib, a third generation EGFR tyrosine kinase inhibitor:current data and future directions. Expert Opin. Pharmacother. 17 (2016) 989–993. DOI:10.1517/14656566.2016.1162786 |
| [11] | M.Z. Qin, T.T. Wang, B.X. Xu, et al., Novel hydrazone moiety-bearing aminopyrimidines as selective inhibitors of epidermal growth factor receptor T790M mutant. Eur. J. Med. Chem. 104 (2015) 115–126. DOI:10.1016/j.ejmech.2015.09.031 |
| [12] | M.Z. Qin, X. Zhai, H.B. Xie, et al., Design and synthesis of novel 2-(4-(2-(dimethylamino)ethyl)-4H-1,2,4-triazol-3-yl)pyridines as potential antitumor agents. Eur. J. Med. Chem. 81 (2014) 47–58. DOI:10.1016/j.ejmech.2014.04.059 |
| [13] | M.Z. Qin, W.K. Liao, C. Xu, et al., Synthesis and biological evaluation of novel 4-(2-fluorophenoxy)-2-(1H-tetrazol-1-yl)pyridines bearing semicarbazone moieties as potent antitumor agents. Arch. Pharm. 346 (2013) 840–850. DOI:10.1002/ardp.v346.11 |
| [14] | W. Zhou, X.F. Liu, Z.C. Tu, et al., Discovery of pteridin-7(8H)-one-based irreversible inhibitors targeting the epidermal growth factor receptor (EGFR) kinase T790M/L858R mutant. J. Med. Chem. 56 (2013) 7821–7837. DOI:10.1021/jm401045n |
| [15] | W.J. Zhou, D. Ercan, L. Chen, et al., Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462 (2009) 1070–1074. DOI:10.1038/nature08622 |