 2017, Vol. 28
2017, Vol. 28 



b University of Chinese Academy of Sciences, Beijing 100049, China;
c National Engineering Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, China
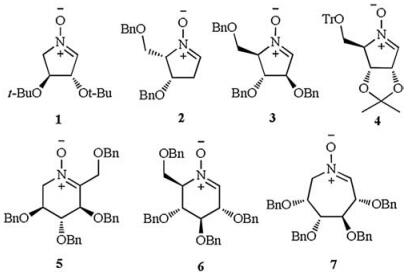
"Nitrogen-in-the-ring" analogues of pyranoses and furanoses, namely iminosugars, azasugars or polyhydroxylated alkaloids, are potent inhibitors of glycosidases and other glycosyl processing enzymes [1, 2]. Therefore, these compounds have great potential in the treatment of type Ⅱ diabetes, cancers and viral infections, and some of them have already been used as drugs, such as NHE-DNJ (Miglitol) and NB-DNJ (Miglustat) [3–6]. Until now, numerous methods have been developed for the synthesis of natural iminosugars and their non-natural analogs from either carbohydrates or non-sugar chiral-pool compounds [7–12]. Among these approaches to iminosugars, polyhydroxylated or sugar-derived cyclic nitrones (1–7, Fig. 1) have been proven to be one of the most useful intermediates due to their capability of undergoing a variety of important transformations, such as 1, 3-dipolar cycloaddition, nucleophilic addition, and pinacol-type coupling reaction [13–15]. Based on our general interests in the synthesis and bioactivity study of iminosugars, we became interested in synthesis of a novel C-branched polyhydroxylated cyclic nitrone, which is likely to be of significant value as a diverse intermediate for the construction of some C-branched pyrrolidine iminosugars with functionalized quaternary centers on the pyrrolidine ring [16–20].

|
Download:
|
| Figure 1. Examples of some polyhydroxylated cyclic nitrones. | |
{kind=link}
Introduction of C-branched chain is a useful method for structural modification of iminosugars in the study of structureactivity relationship, and some of these alkaloids have already been reported (8–16, Fig. 2). For example, isoDAB (8) was a potent but more selective α-glucosidase inhibitor comparing to its parent iminosugar DAB [21]. 4-C-Me-DAB (9) is a competitive, specific and potent α-glucosidase inhibitior [22]. Compound 10 shows potent inhibition to α-glucosidase from rice (IC50 = 5 μmol/L), while L-isoDMDP (11) is potent specific competitive inhibitor of gut disaccharidases (Ki = 81 mmol/L for rat intestinal maltase) and is more effective in the suppression of hyperglycaemia in a maltose loading test than Miglitol [23, 24]. Both compound 12 and 13 display moderate inhibitory activity against Mycobacterium smegmatis galactan biosynthesis [25]. Compound 14 was a specific potent α-glucosidase inhibitor (IC50 = 52 nmol/L, rice α-glucosidase), while DNJ and castanospermine are also inhibitors of β-glucosidase [15]. Azepane 15 displays selective and moderate competitive L-fucosidase inhibition [26]. Early in this year, a new iminosugar 16 [27] had been synthesized from D-glucose by Zhang et al., whose enantiomer could be synthesied from the title cyclic nitrone in one step.

|
Download:
|
| Figure 2. Structures of some branched iminosugars. | |
{kind=link}
2. Results and discussion
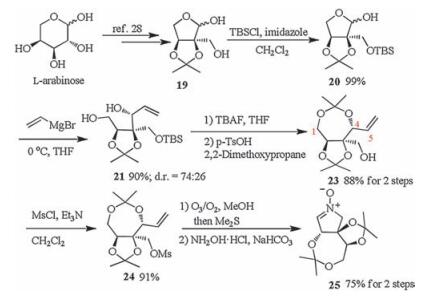
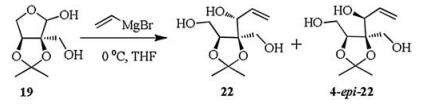
The synthetic route of the title nitrone was shown in Scheme 1. (2S, 3S)-2-Hydroxymethyl-2, 3-O-isopropylidene-L-erythrose (19) was prepared in 74% yield from commercially available L-arabinose according to the literature reported method [28]. The detail is deposited in Supporting information. Initially, we attempted to obtain the triol 22 by the Grignard addition between compound 19 and vinyl magnesium bromide without supplementing more protecting groups (Scheme 2). Unfortunately, the resulting two diastereomers 22 and 4-epi-22 are a pair of inseparable isomers and failed to be separated by flash column chromatography.

|
Download:
|
| Scheme 1. Synthesis of (2S, 3R, 4R)-1-amino-1, 4-anhydro-1, N-didehydro-3-(1, 2-dihydroxyl)ethyl-2, 6:3, 5-di-O-isopropylidene-L-threose N-oxide (25). | |
{kind=link}

|
Download:
|
| Scheme 2. Attempt of the synthesis of triol 22. | |
{kind=link}
Thus, compound 19 was first transformed to silyl ether 20 and then underwent Grignard addition to produce diol 21 (d.r. = 74:26), the C4 configuration of which was confirmed as R by the crystal structure of compound 25. The TBS protecting group of diol 21 was removed by TBAF, and then the C1 and C5 hydroxyl groups were protected by acetonide to give alcohol 23. The free hydroxyl group in 23 was activated by mesyl chloride to afford methanesulfonate 24. Compound 24 was then ozonized to give the intermediate aldehyde, which was used directly due to its instability. The aldehyde was then put under routine procedure to give the title nitrone, (2S, 3R, 4R)-L-amino-1, 4-anhydro-1, N-didehydro-3-(1, 2-dihydroxyl)ethyl-2, 6:3, 5-di-O-isopropylidene-l-threose N-oxide (25). Compound 25 is unstable and would decompose while purified by silica gel column chromatography. However, it was found stable when purified by alkaline aluminum oxide column chromatography [29]. The structure of compound 25 was confirmed unambiguously by spectroscopic data and X-ray crystal structure (Fig. 3). The crystal data is deposited in Supporting information and at the Cambridge Crystallographic Data Centre with number of CCDC 1515829.

|
Download:
|
| Figure 3. Crystal structure of compound 25. | |
{kind=link}
3. Conclusion
In summary, a novel C-branched polyhydroxylated cyclic nitrone 25 with a quaternary chiral center has been synthesized starting from commercially available L-arabinose in 29.0% total yield, with Grignard addition, ozonization and cyclization as the key steps.
4. Experimental 4.1. General methodsNMR spectra was recorded at 300 MHz or 400 MHz (1H NMR) and 75 MHz, 100 MHz (13C NMR) in CDCl3 (with TMS as internal standard). High-resolution mass spectra (HRMS) were performed with a LTQ/FT linear ion trap mass spectrometer. All reagents were used as received from commercial sources without further purification or prepared as described in the literature. Tetrahydrofuran was distilled from sodium and benzophenone. TLC plates were visualized by treatment with a spray of Pancaldi reagent ((NH4)6MoO4, Ce(SO4)2, H2SO4, H2O). Polarimetry was determined using a polarimeter with concentrations (c) given in gram per 100 mL.
4.2. Synthesis of (2S, 3S)-2-((tert-butyldimethylsilyl)oxy)methyl-2, 3-O-isopropylidene-L-erythrose (20)TBSCl (10.1 g, 67.1 mmol) and imidazole (6.1 g, 89.4 mmol) were added into a solution of compound 16 (8.5 g, 44.7 mmol) in dichloromethane (150 mL) and the reaction mixture was stirred at room temperature for 6 h. Then water (100 mL) was added, the aqueous and organic phases were separated and the aqueous phase was extracted with dichloromethane (100 mL × 3). The organic phases were combined, dried with MgSO4, concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (petroleum ether/EtOAc = 5/1) to give compound 17 as a light yellow syrup (13.4 g, 99%). Data for 17: [α]D20 +11 (c 1.05 in CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 5.33– 5.31 (d, 0.5H, J = 6.6 Hz), 4.98–4.94 (d, 0.5H, J = 12 Hz), 4.67–4.66 (d, 1H, J = 3 Hz), 4.67–4.66 (d, 1H, J = 3 Hz), 4.38–4.36 (d, 0.5H, J = 6.6 Hz), 4.08–3.94 (m, 2H), 3.86–3.74 (m, 2H), 3.56–3.51 (m, 0.5H), 1.55 (s, 1.5H), 1.48 (s, 1.5H), 1.43 (s, 1.5H), 1.39 (s, 1.5H), 0.92 (s, 4.5H), 0.90 (s, 4.5H), 0.13 (s, 3H), 0.09 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 113.50, 113.42, 104.75, 97.72, 92.19, 88.30, 83.34, 82.77, 72.09, 68.03, 64.16, 63.09, 27.48, 27.45, 27.05, 26.77, 25.76, 25.74, 18.14, -5.59, -5.64, -5.68; HRMS(ESI) calcd. for C14H28O5SiNa+ [M +Na]+ 327.1598, found 327.1593.
4.3. Synthesis of (2S, 3R, 4R)-2-((tert-butyldimethylsilyl)oxy)methyl-2, 3-O-isopropylidene-5-ene-1, 4-diol (21)Compound 20 (100.0 mg, 0.3 mmol) was dissolved in anhydrous THF (10 mL), and vinyl magnesium bromide (1 mol/L, 0.3 mL) was added dropwise at 0 ℃ under Ar atmosphere. The reaction mixture was stirred for 1 h, and then saturated aqueous NH4Cl solution was added dropwise to quench the reaction. Water (10 mL) was added, the organic and the aqueous phases were separated and the aqueous phase was extracted with ethyl acetate (20 mL × 3). The organic phases were combined, dried with MgSO4, concentrated under reduced pressure and the crude product was purified by column chromatography on silica gel (petroleum ether/EtOAc = 3/ 1) to give compound 21 as a light yellow syrup (21, 73.1 mg: 4-epi-21, 25.7 mg; 90% total yeild). Data for 21: [α]D20 +52 (c 2.15 in CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 6.17–6.10 (m, 1H), 5.56–5.28 (m, 2H), 4.41–4.37 (m, 1H), 4.30–4.27 (m, 1H), 4.09–3.94 (m, 3H), 3.77–3.75 (m, 1H), 3.63–3.60 (m, 1H), 3.34–3.30 (m, 1H) 1.45 (s, 3H), 1.37 (s, 3H), 0.90 (s, 9H), 0.09 (s, 6H); 13C NMR (75 MHz, CDCl3): d 136.88, 115.87, 108.40, 82.40, 81.42, 73.24, 65.57, 61.06, 28.46, 26.76, 25.78, 18.13, -5.67, -5.83; HRMS(ESI) calcd. for C16H32O5SiNa+ [M+Na]+ 355.1911, found 355.1906.
4.4. Synthesis of (2S, 3R, 4R)-2-hydroxymethyl-1, 4:2, 3-di-Oisopropylidene-5-hexene (23)Compound 21 (1.0 g, 3.0 mmol) and TBAF (950.0 mg, 3.0 mmol) were dissolved in THF (25 mL), and the reaction mixture was stirred at room temperature for 2 h. Water (10 mL) was added and the aqueous phase was extracted with ethyl acetate (40 mL × 7). The organic phases were combined, dried with MgSO4, concentrated under reduced pressure and the crude product was used in the next step without further purification. The crude product, 2, 2-dimethyoxypropane (376.4 mg 3.6 mmol) and p-TSA (51.6 mg, 0.3 mmol) were dissolved in DMF (10 mL), and the reaction mixture was stirred at r.t. for 10 h. H2O (30 mL) was added and the aqueous phase was extracted with ethyl acetate (30 mL × 3). The organic phases were combined, dried with MgSO4, the solvent was removed under reduced pressure and the crude product was purified by column chromatography on silica gel (petroleum ether/ EtOAc = 4/1) to give compound 23 as a yellow syrup (684.2 mg, 88% for 2 steps). Data for 23: [a]D20 +79 (c 1.75 in CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 6.07–5.97 (m, 1H), 5.39–5.23 (m, 2H), 4.55 (dd, 1H, J = 3.5, 1.9 Hz), 4.14 (s, 1H), 4.00 (d, 2H, J = 1.5 Hz), 3.78–3.60 (m, 2H), 2.13–2.08 (m, 1H), 1.55 (s, 3H), 1.40 (s, 6H), 1.32 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 133.84, 116.07, 108.01, 101.86, 83.43, 77.45, 77.38, 77.03, 76.60, 71.30, 61.36, 58.95, 28.55, 26.13, 24.27, 23.51; HRMS(ESI) calcd. for C13H22O5Na+ [M+Na]+ 281.1359, found 281.1360.
4.5. Synthesis of (2S, 3R, 4R)-2-((mesyloxy)methyl)-1, 4:2, 3-di-Oisopropylidene-5-hexene (24)Compound 23 (54.0 mg, 0.21 mmol) and Et3N (31.7 mg, 0.31 mmol) were dissolved in dichloromethane (25 mL), and mesyl chloride (28.8 mg, 0.25 mmol) at 0 ℃. The reaction mixture was stirred at room temperature for 8 h, water (20 mL) was added. The organic and the aqueous were separated and the aqueous phase was extracted with dichloromethane (20 mL × 3). The organic phases were combined, dried with MgSO4, concentrated under reduced pressure and the crude product was purified by column chromatography on silica gel (petroleum ether/EtOAc = 4/1) to give compound 24 as a colorless syrup (64.0 mg, 91%). Data for 24: [a]D20 79 (c 6.60 in CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 6.07–5.97 (m, 1H), 5.42–5.25 (m, 2H), 4.52–4.51 (m, 1H), 4.40–4.25 (m, 2H), 4.13 (s, 1H), 3.98 (s, 2H), 3.02 (s, 3H), 1.55 (s, 3H), 1.41 (s, 3H), 1.40 (s, 3H), 1.31 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 132.86, 116.76, 108.31, 101.92, 82.74, 75.53, 70.38, 66.99, 58.18, 37.41, 28.42, 25.83, 24.26, 23.51; HRMS(ESI) calcd. for C14H24O7SNa+ [M+Na]+ 359.1135, found 359.1131.
4.6. Synthesis of (2S, 3R, 4R)-1-amino-1, 4-anhydro-1, N-didehydro-3-(1, 2-dihydroxyl)ethyl-2, 6:3, 5-di-O-isopropylidene-L-threose N-oxide (25)The solution of compound 24 (44.0 g, 0.13 mmol) in CH3OH (20 mL) was cooled to -60 ℃ and purged with oxygen, then submitted to the ozonization procedure until TLC showed completion of the reaction. The reaction mixture was purged with Ar and quenched by dimethyl sulfide. The resulting mixture was stirred at room temperature for 2 h, and then concentrated in vacuo. Water (20 mL) was added and the aqueous phase was extracted with ethyl acetate (20 mL × 3). The organic phases were combined, dried with MgSO4, concentrated under reduced pressure and the residue was used without further purification. The crude product was dissolved in ethanol (8 mL) and H2O (2 mL), NH2OH·HCl (27 mg, 0.39 mmol) and NaHCO3 (44 mg, 0.52 mmol) were added, and the reaction mixture was stirred at r.t. for 8 h. Then H2O (20 mL) was added and the aqueous phase was extracted with ethyl acetate (20 mL × 3). The organic phases were combined, dried with MgSO4, the solvent was removed under reduced pressure and the crude product was purified by column chromatography on alkaline aluminum oxide (petroleum ether/ EtOAc = 1/1) to give compound 25 as a white solid (25 mg, 75.0% for 2 steps). Data for 25: mp: 56–57 ℃; [a]D20 +56 (c 1.80 in CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 6.89 (s, 1H), 4, 794–4.790 (d, 1H, J = 1.6 Hz), 4.33–4.29 (m, 1H), 4.14 (s, 1H), 4.07–4.03 (m, 1H), 3.93–3.81 (m, 2H), 1.46 (s, 3H), 1.39 (s, 3H), 1.38 (s, 3H), 1.37 (s, 3H); 13C NMR (100 MHz, CDCl3): d 132.25, 109.29, 102.26, 83.94, 78.28, 77.59, 69.94, 57.99, 27.60, 25.28, 24.37, 24.00; HRMS(ESI) calcd. for C12H20O5+ [M+H]+ 258.1336, found 258.1334.
AcknowledgmentsFinancial support from National Basic Research Program of China (No. 2012CB822101), the National Natural Science Foundation of China (No. 21272240), National Science and Technology Major Projects for "Major New Drugs Innovation and Development" (No. 2013ZX09508104) and National Engineering Research Center for Carbohydrate Synthesis of Jiangxi Normal University are gratefully acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.12.033.
| [1] | N. Asano, R.J. Nash, R.J. Molyneux, G.W.J Fleet, Sugar-mimic glycosidase inhibitors:natural occurrence biological activity and prospects for therapeutic application. Tetrahedron:Asymmetry 11 (2000) 1645–1680. DOI:10.1016/S0957-4166(00)00113-0 |
| [2] | A.A. Watson, G.W.J. Fleet, N. Asano, R.J. Molyneux, R.J Nash, Polyhydroxylated alkaloids-natural occurrence and therapeutic applications. Phytochemistry 56 (2001) 265–295. DOI:10.1016/S0031-9422(00)00451-9 |
| [3] | C. Decroocq, F. Stauffert, O. Pamlard, Iminosugars as a new class of cholinesterase inhibitors. Bioorg.Med.Chem.Lett. 25 (2015) 830–833. DOI:10.1016/j.bmcl.2014.12.071 |
| [4] | H. Chen, L. Hao, M. Zhu, Synthesis of bi-/tricyclic azasugars fused thiazinan-4-one and their HIV-RT inhibitory activity. Bioorg.Med.Chem.Lett. 24 (2014) 3426–3429. DOI:10.1016/j.bmcl.2014.05.079 |
| [5] | R. Lahiri, A.A. Ansari, Y.D Vankar, Recent developments in design and synthesis of bicyclic azasugars, carbasugars and related molecules as glycosidase inhibitors. Chem.Soc.Rev. 42 (2013) 5102–5118. DOI:10.1039/c3cs35525j |
| [6] | C.H. Hill, A.H. Viuff, S.J. Spratley, Azasugar inhibitors as pharmacological chaperones for Krabbe disease. Chem.Sci. 6 (2015) 3075–3086. DOI:10.1039/C5SC00754B |
| [7] | Q. Li, X.S Ye, Iminosugars as immunomodulating agents:synthesis and biological activities of 1-deoxynojirimycin and related compounds. Isr.J. Chem. 55 (2015) 336–346. DOI:10.1002/ijch.v55.3/4 |
| [8] | H Takahata, Synthetic medicinal chemistry of the biomolecular components mimics. Yakugaku Zasshi 133 (2013) 575–585. DOI:10.1248/yakushi.13-00054 |
| [9] | H Takahata, Chiral synthesis of iminosugars. Heterocycles 85 (2012) 1351–1376. DOI:10.3987/REV-12-734 |
| [10] | A.L. Concia, L. Gomez, T. Parella, J. Joglar, P Clapes, Casuarine stereoisomers from achiral substrates:chemoenzymatic synthesis and inhibitory properties. J.Org.Chem. 79 (2014) 5386–5389. DOI:10.1021/jo500991p |
| [11] | C.K. Lin, L.W. Cheng, H.Y. Li, W.Y. Yun, W.C Cheng, Synthesis of novel polyhydroxylated pyrrolidine-triazole/-isoxazole hybrid molecules. Org. Biomol.Chem. 13 (2015) 2100–2107. DOI:10.1039/C4OB01934B |
| [12] | C.M. Si, Z.Y. Mao, H.Q. Dong, Divergent method to trans-5-hydroxy-6-alkynyl/alkenyl-2-piperidinones:syntheses of (-)-epiquinamide and (+)-swainsonine. J.Org.Chem. 80 (2015) 5824–5833. DOI:10.1021/acs.joc.5b00803 |
| [13] | D. Martella, G. D'Adamio, C. Parmeggiani, Cycloadditions of sugar-derived nitrones targeting polyhydroxylated indolizidines. Eur.J.Org.Chem. (2016) 1588–1598. |
| [14] | M. Soluch, B. Grzeszczyk, O. Staszewska-Krajewska, M. Chmielewski, B. Furman, Synthesis of thienamycin methyl ester from 2-deoxy-D-ribose via Kinugasa reaction. J.Antibiot. 69 (2016) 164–168. DOI:10.1038/ja.2015.108 |
| [15] | J. Boisson, A. Thomasset, E. Racine, Hydroxymethyl-branched polyhydroxylated indolizidines:novel selective a-glucosidase inhibitors. Org.Lett. 17 (2015) 3662–3665. DOI:10.1021/acs.orglett.5b01505 |
| [16] | Y.Y. Song, K. Kinami, A. Kato, First total synthesis of(+)-broussonetine W: glycosidase inhibition of natural product&analogs. Org.Biomol.Chem. 14 (2016) 5157–5174. DOI:10.1039/C6OB00720A |
| [17] | B.C. Qian, A. Kamori, K. Kinami, Epimerization of C5 of an N-hydroxypyrrolidine in the synthesis of swainsonine related iminosugars. Org. Biomol.Chem. 14 (2016) 4488–4498. DOI:10.1039/C6OB00531D |
| [18] | Y.X. Li, R. Iwaki, A. Kato, Fluorinated radicamine A and B:synthesis and glycosidase inhibition. Eur.J.Org.Chem. (2016) 1429–1438. |
| [19] | J.S. Zhu, S. Nakagawa, W. Chen, Synthesis of eight stereoisomers of pochonicine:nanomolar inhibition of beta-N-acetylhexosaminidases. J.Org. Chem. 78 (2013) 10298–10309. DOI:10.1021/jo401694e |
| [20] | W. Zhang, K. Sato, A. Kato, Synthesis of fully substituted polyhydroxylated pyrrolizidines via cope-house cyclization. Org.Lett 13 (2011) 4414–4417. DOI:10.1021/ol201749c |
| [21] | D. Best, S.F. Jenkinson, A.W. Saville, Cystic fibrosis and diabetes:isoLAB and isoDAB enantiomeric carbon-branched pyrrolidine iminosugars. Tetrahedron Lett. 51 (2010) 4170–4174. DOI:10.1016/j.tetlet.2010.05.131 |
| [22] | F.P.d. Cruz, S. Newberry, S.F. Jenkinson, 4-C-Me-DAB and 4-C-Me-LAB— enantiomeric alkyl-branched pyrrolidine iminosugars—are specific and potent a-glucosidase inhibitors; acetone as the sole protecting group. Tetrahedron Lett. 52 (2011) 219–223. DOI:10.1016/j.tetlet.2010.10.173 |
| [23] | N.J. Pawar, V.S. Parihar, S.T. Chavan, α-Geminal dihydroxymethyl piperidine and pyrrolidine iminosugars:synthesis, conformational analysis, glycosidase inhibitory activity, and molecular docking studies. J.Org.Chem. 77 (2012) 7873–7882. DOI:10.1021/jo3009534 |
| [24] | S.F. Jenkinson, D. Best, A.W. Saville, C-branched iminosugars:alpha-glucosidase inhibition by enantiomers of isoDMDP, isoDGDP, and isoDAB-L-isoDMDP compared to miglitol and miglustat. J.Org.Chem. 78 (2013) 7380–7397. DOI:10.1021/jo4005487 |
| [25] | S. Cren, C. Wilson, N.R Thomas, A rapid synthesis of hexofuranose-like iminosugars using ring-closing metathesis. Org.Lett. 7 (2005) 3521–3523. DOI:10.1021/ol051232b |
| [26] | H. Li, Y. Zhang, S. Favre, Synthesis of branched seven-membered 1-N-iminosugars and their evaluation as glycosidase inhibitors. Carbohydr.Res. 356 (2012) 110–114. DOI:10.1016/j.carres.2011.10.039 |
| [27] | E. Zhang, P.Y. Bai, W. Sun, Synthesis and glycosidase inhibition evaluation of(3S, 4S)-3-((R)-1, 2-dihydroxyethyl)pyrrolidine-3, 4-diol. Carbohydr.Res. 434 (2016) 33–36. DOI:10.1016/j.carres.2016.08.003 |
| [28] | ${referAuthorVo.mingEn} F.León, I. Brouard, A. Rivera, Isolation, structure elucidation, total synthesis, and evaluation of new natural and synthetic ceramides on human SK-MEL-1 melanoma cells. J.Med.Chem. 49 (2006) 5830–5839. DOI:10.1021/jm0605334 |
| [29] | W.B. Wang, M.H. Huang, Y.X. Li, A practical synthesis of sugar-derived cyclic nitrones:powerful synthons for the synthesis of iminosugars. Synlett (2010) 488–492. |