 2017, Vol. 28
2017, Vol. 28 



b Petro China Company Limited, Beijing 100007, China
Hydrogels, as one of the most attractive soft materials, have been extensively studied due to their versatility and widespread applications, such as drug and cell carriers, tissue engineering matrices, etc.[1-3]. Conventionally, hydrogels are flexible and have a rather low strength so that they can be easily damaged by cracks or totally broken under weak external force, which is the sever obstacle to their safe and long-time application [4, 5]. To solve this problem, the concept of self-healing hydrogel, i.e. the hydrogel can rapidly repair the splits or fracture surfaces automatically once the damage occurs, has been proposed and quickly attracted intensive interest of the material scientists [6, 7]. Currently, the driven forces to the self-healing behavior in materials can be originated from hydrogen bonds [8, 9], hydrophobic interactions [10, 11], reversible chemical bonds [12-14], and so on [15-17]. For example, Waite et al. synthesized polyacrylate and polymethacrylate materials surface-functionalized with mussel-inspired catechols. The extensive catechol-mediated interfacial hydrogen bonds enabled the materials to self-heal once they were broken underwater [18]. The widely-used polyacrylamide (PAM)-based gels also need the selfhealing feature in their various applications such as removal of heavy metal ions and enhanced oil recovery [19, 20]. Okay et al. prepared PAM hydrogel via the copolymerization of acrylamide (AM) and stearyl methacrylate (C18) or dococyl acrylate (C22) in sodium dodecyl sulfate (SDS) micelles, which exhibited high selfhealing efficiency through the dynamic nature of the junction zones between the network chains consisting of C18 or C22 [21].
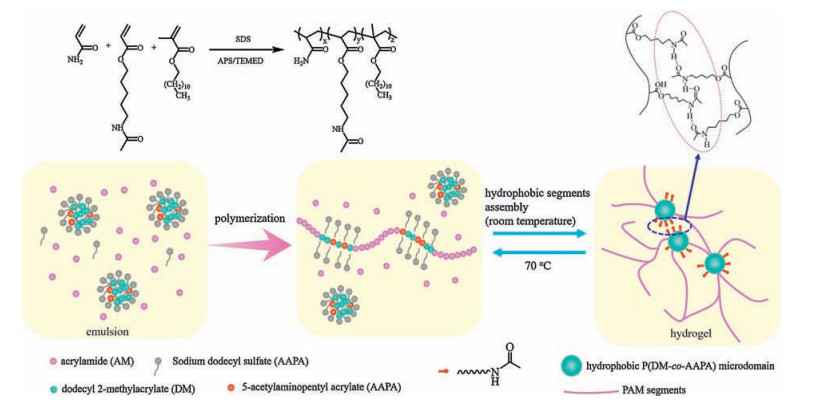
In this study, we proposed a facile synthesis method to prepare a strong and self-healable PAM-based hydrogel (sHG) via a onestep emulsion copolymer of AM and the other two functional monomers (dodecyl 2-methacrylate (DM) and 5-acetylaminopentyl acrylate (AAPA)). The crosslinks in the obtained strong hydrogel are hydrophobic microdomains which were spontaneously formed and separated during the emulsion copolymerization. As a result, the obtained hydrogel can achieve reversible sol-gel transition triggered by the environment temperature. Furthermore, the prepared hydrogels have excellent self-healing ability mainly based on the hydrogen bonds formed by the amide bonds in the segments constituted by AAPA units.
2. Results and discussion 2.1. Synthesis of self-healable PAM-based hydrogel (sHG) via emulsion copolymerizationThe macromolecular chain structures formed by the emulsion copolymerization of hydrophilic and hydrophobic monomers have been widely studied in literatures [22-24]. It had been reported that multi-block polymer chains will be formed when the number of hydrophobic monomers in per surfactant micelle (NH) is higher than 1 [25]. NH can be calculated by the following Eq. (1) [26]:
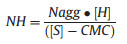
|
(1) |
Where Nagg is the aggregation number of the surfactant, i.e. the number of micelle. [S] and CMC are the actual molar concentration and the critical micellar concentration of the surfactant, respectively. [H] is the molar concentration of hydrophobic monomer. Based on the above theory, a multi-block polymer chains constituted of hydrophilic PAM segments and hydrophobic P (DM-co-AAPA) segments were designed to be prepared via emulsion copolymerization. Here, SDS was chosen as the surfactant. Thus, CMC is 8 × 10-3 mol/L, [SDS] was fixed as 0.243 mol/L, [DM + AAPA] was controlled within 0.007-0.041 mol/L, and Nagg is 57 at the polymerization temperature of 25 ℃ [27]. According to Eq. (1), the NH in our system would be 1.64-9.94, much higher than 1. In practice, the emulsion copolymerization of AM, DM, and AAPA of all investigated recipes were so fast at room temperature that the whole reaction system transformed into a gel within several minutes, as shown in Fig. 1.

|
Download:
|
| Figure 1. The digital photos of the sHG samples prepared via the emulsion copolymerization of AM, DM, and AAPA at room temperature initiated by APS and TEMED. | |
The crosslinking mechanism to form a gel should be related with the phase separation of hydrophobic segments and the generation of hydrogen bonds between AAPA units on copolymer chains as reported in literatures [28, 29]. The structure of multiblock copolymer chains and the formation mechanism of sHG are illustrated in Scheme 1.

|
Download:
|
| Scheme 1. The structure of multi-block copolymer chains formed by the emulsion copolymerization of AM, DM, and AAPA, and the formation mechanism of thermalreversible sHG. | |
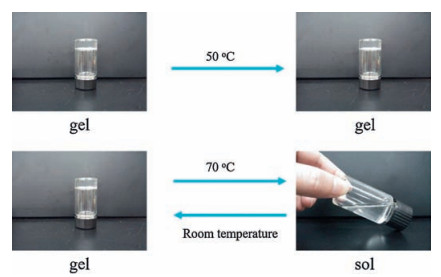
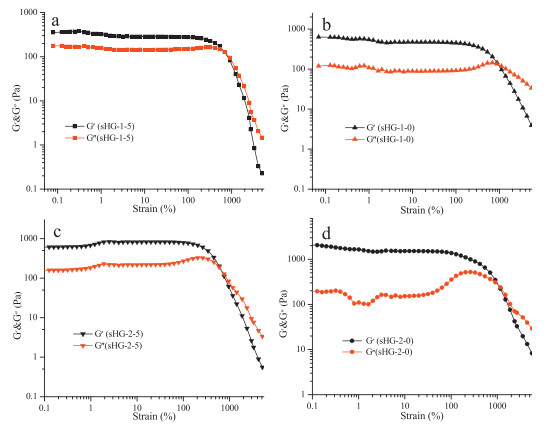
Initially, the polymerization of AM in aqueous continuous phase was initiated by APS and TEMED. When the growing AM oligomer chain radical encounters a monomer-swollen micelle, the copolymerization of hydrophobic monomer units will be initiated in the micelle to form a hydrophobic block, till the growing chain radical encounters AM molecules and continues to form another PAM segment in the aqueous solution. The above chain growth processes repeat, finally a multi-block structured polymer chain forms [25]. During the polymerization process, the increasing hydrophobic segments will self-assemble driven by the hydrophobic interactions and form insoluble microdomains to separate from water. These isolated domains act as the crosslinks of all the copolymer chains, resulting in the formation of a three dimensional polymer network. The storage modulus G' and the loss modulus G" of the sHG samples containing different contents of DM and AAPA are shown in Fig. 2a and 2c, respectively. It can be seen that in the high frequency range (>1 rad/s), the storage moduli (G') of all sHG samples exceed the corresponding loss moduli (G"), indicating that the gels are elastic. While in the low frequency, the G"s are larger than the G's, implying a liquid-like response. According to the time-temperature equivalence principle, it can be expected that when the environment temperature increase above a certain value, these gels will be transformed into liquid. A typical temperature-dependent sol-gel transition is shown in Fig. 3. When the gel was heated to 50 ℃, there is no change in the appearance. However, when the temperature continued to rise to 70 ℃, the gel transformed into a sol state. The sol state also can revert back to a gel state during the cooling process. These thermal-responsive reversible sol-gel transition proves that the crosslinks in the gel are formed through physical interactions, not chemical bonds. We know that the non-chemical crosslinked amorphous polymer chain microdomains will be melt or dissolved at high temperature. Therefore, the above results confirm that the phase separation of hydrophobic segments plays the key role to the formation of sHG.

|
Download:
|
| Figure 2. Storage modulus G', loss modulus G"(a, c) and loss tangent (tanδ) (b, d) of sHG samples as a function of frequency | |

|
Download:
|
| Figure 3. The digital photos of sol-gel transition of sHG-1-5 at 50 ℃ and 70 ℃. | |
It is also noted in Fig. 2a and c that the viscoelasticity of the prepared sHGs has a weak dependence on the content of AAPA. Under the same content of DM, G' of the gels have a weak increase with the increase of the content of AAPA, but G" the reverse. Evidently, the hydrophobicity of AAPA is less than DM due to the polar end amide bond. The hydrophobic interactions between the P (DM-co-AAPA) will be weakened with the increase of AAPA units, resulting in the decrease of G' and the increase of G". The loss tangent (tanδ) in Fig. 2b and d show the same tendency. In a board range of frequencies, nearly all tanδ are lower than 1, indicating a hydrogel texture of all samples [30, 31]. But at very low frequencies, tanδ is larger than 1, suggesting that a sol state exists.
On the contrary, G' and G" of sHG also have a great dependence on the content of DM, as compared with Fig. 2a and c. G' will be greatly improved with the increase of the DM content, and G" the reverse. For example, G' rises up from 400 Pa for sHG-1-5 to 700 Pa for sHG-2-5 at the same frequency of 10 rad/s, while the G" decreases from 200 Pa to 150 Pa. It indicates that the strength of the sHG is mainly determined by the content of DM.
2.2. Self-healing performance of sHGsTo assess the self-healing ability of the prepared sHGs, the rheological recovery test of the sample sHG-1-5 were firstly performed. The strain-sweep test of sHG-1-5 in a constant frequency of 6.28 rad/s over the strain range of 0.1-5000 at 25 ℃ are presented in Fig. 4a, compared with those of sHG-1-0 (Fig. 4b). It can be seen that the storage modulus G' of both sHG-1-5 and sHG-1-0 are larger than the corresponding loss modulus G"s. The difference between G' and G" keeps constant under the small strain ( < 100%), indicating that the hydrogel network is stable and without apparent damaged. However, for sHG-1-5, when the strain is over 600%, G' becomes smaller than G". At the same time, both G' and G" sharply drop, indicating a destruction of hydrogel network or physical cross-linking points [11, 32]. As to the sample sHG-1-0, the destruction occurs at a relatively high strain value (>880%), indicating the strength of sHG-1-0 is higher than sHG-1-5, which is in accord with the result in Fig. 2a. With the increase of the DM content, the transformations of sHG-2-5 and sHG-2-0 occur at strain 700% and 1000% respectively (Fig. 4c and d). When the strain is over 1000%, sHG-2-0 will be completely crushed and cannot be repaired.

|
Download:
|
| Figure 4. Storage modulus G' and loss modulus G" as a function of strain of sHG-1-5 (a), sHG-1-0 (b), sHG-2-5 (c), and sHG-2-0 (d). | |
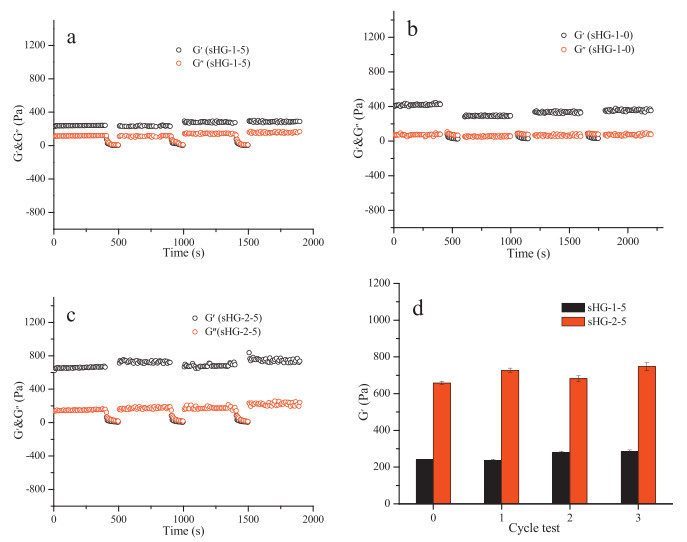
Cyclic test was implemented with toggling strain between 1% and 1400% to measure the self-healing performance of the sHG in a constant frequency of 6.28 rad/s. As shown in Fig. 5, first 1% strain was conducted, the G' and G" remain constant within 400 s, and the G' is larger than G", indicating that the hydrogel is stable and without apparent destroy. When the strain was increased to 1400% and kept for 100 s, the G' and G" fall dramatically, but the G' is a little smaller than G", demonstrating the damage of the hydrogel's network. After two minutes, the strain was reverted from 1400% back to 1%, both G' and G" of sHG-1-5 recover to the original level (Fig. 5a), meaning an almost 100% of self-healing efficiency. However, as for sHG-1-0, both G' and G" can only recover to about 75% of their original state (Fig. 5b). Fig. 6 visually shows the rapid and efficient self-healing performance of sHG-1-5. An original whole gel stick of sHG-1-5 was cut into four pieces. Put the broken pieces together and make them be contacted with each other. Two minutes later, the broken pieces reunited into a new whole stick without any external force. The stick still exhibits excellent tensile property.

|
Download:
|
| Figure 5. Time-sweep curves of G' and G" and test cycles of sHG-1-5 (a), sHG-1-0 (b), sHG-2-5 (c) and the variation of G' of sHG-1-5 and sHG-2-5 after test cycles. | |

|
Download:
|
| Figure 6. The rapid self-healing performance of sHG-1-5. | |
The same excellent self-healing efficiency can be achieved when the DM content is raised. Fig. 5c and 5d show that the G' and G" of sHG-2-5 also can recover 100% to the original level, even become a little higher after the cycle tests. The result is so exciting because although multi-block polymer chains constituted by hydrophilic PAM segments and other hydrophobic segments had been reported in literatures, no self-healing ability can be achieved when the content of hydrophobic monomer is over 2% [21, 33]. Evidently, the excellent self-healing performance of our prepared sHG should be attributed to the introduction of the monomer AAPA, which can produce strong hydrogen interactions through the amide bonds on the end of the long aliphatic chain, as illustrated in Scheme 1. Although the introduction of AAPA weakens the hydrophobic interaction between the hydrophobic segments, the strong intermolecular and intramolecular hydrogen bonding formed by AAPA and AM could enhance the strength of the crosslinks to a certain degree, but more importantly, endows the gel with the fantastic self-healing ability.
3. ConclusionIn summary, self-healable polyacrylamide-based hydrogels were one-step prepared via the emulsion copolymerization of AM, DM, and AAPA using SDS as the emulsifier at room temperature. The produced linear multi-block copolymer chains constituted by hydrophilic PAM segments and hydrophobic P (DMco-AAPA) segments. The P (DM-co-AAPA) segments self-aggregated into hydrophobic microdomains forced by the hydrophobic interactions, and finally be separated from water phase, acting as the crosslinks and leading to the formation of strong hydrogels with a storage modulus as high as 400 Pa. These hydrophobic microdomains will be dissolved in water when the temperature increases to 70 ℃, indicating a temperature-responsive reversible sol-gel transition of the prepared hydrogels. The introduction of a little amount of AAPA endows the prepared gels with excellent self-healing ability. The broken hydrogels can be automatically healed into a body with a same strength within 2-min's contact. This work provides a new and facile way to prepare thermalresponsive reversible hydrogels with high strength and selfhealing efficiency.
4. Experimental 4.1. Materials5-Amino-1-pentanol (97%), ethyldiisopropylamine (DIPEA, 99%), and dodecyl 2-methacrylate (DM, 96%) were purchased from Aladdin Industrial Corporation. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl, 99%) was supplied by Energy Chemical Reagent Co., Ltd. Acrylic acid (AA, AR) were supplied by Sinopharm Chemical Reagent Co., Ltd., and purified by vacuum distillation. Sodium dodecyl sulfate (SDS, CP), acrylamide (AM, CP), ammonium persulfate (APS, AR), N, N, N', N'-tetramethylethylenediamine (TEMED, BR), and other analytical pure chemicals were purchased from Sinopharm Chemical Reagent Co., Ltd. and used as received. Deionized water was utilized in all experiments. APS and TEMED stock solutions were respectively prepared by dissolving 0.8 g of APS and 0.25 mL of TEMED in 10 mL of water.
4.2. Preparation of 5-acetylaminopentyl acrylate (AAPA)AAPA was synthesized according to the literature [34], as illustrated in Scheme 2.

|
Download:
|
| Scheme 2. The synthetic route of AAPA | |
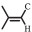
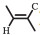
5-Amino-1-pentanol (2.6 mL, 25 mmol) was dissolved in ethyl acetate (50 mL), followed by the addition of acetic anhydride (2.58 mL, 27.5 mmol) dropwise. After being stirred for 3 h at room temperature, the mixture was vacuum dried at room temperature to evaporate the solvent. The dried product (1.5 g, 10.3 mmol), AA (1.12 g, 15.4 mmol), DIPEA (2.8 mL, 17 mmol), EDC·HCl (3.27 g, 17 mmol) were added in dichloromethane (DCM, 50 mL). After being stirred for 24 h at room temperature, the mixture was extracted with 1 mol/L NaOH (50 mL), 1 mol/L HCl (50 mL), saturated NaHCO3 (50 mL), and brine (50 mL) in order, finally dried with MgSO4 overnight. The clear solution was filtered and the solvent was evaporated by reduce pressure. The obtained crude product was purified by flash column chromatography using DCM: CHCl3 (1:1) as the eluent. The 1H NMR spectrum of the purified product, AAPA, was measured on Bruker AV 300 MHz spectrometer using the DMSO-d6 as the solvent, as shown in Fig. 7. All the peaks can be assigned to all the hydrogen atoms in the structure of AAPA (δ: 7.78 (s, H, -NH-), 6.31 (m, H, 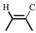

|
Download:
|
| Figure 7. The 1H NMR spectrum of AAPA | |
4.3. Preparation of self-healing PAM-based hydrogel (sHG)
SDS (0.7g) was dissolved in 10mL of water. DM and AAPA were added at a various ratio and stirred for 1h. After purging with N2 for 5min, AM, TEMED, and APS were added in. The whole system rapidly transformed into a gelatinous texture within several minutes. The system was sealed and stood for 12h at room temperature in order to guarantee the reaction to be finished as completelyas possible. The detailed recipes for all the sHG samples are listed in Table 1. To prepare the hydrogel samples for the rheological measurement, the above mentioned reaction solutions were poured into the cylindrical space with a height of 2mm fenced by a circular rubber band sandwiched between two glass plates.
|
|
Table 1 The recipes for the prepared sHG samples. ([SDS]=7w/v %; [APS]=3.5mmol/L;[TEMED]=16.7mmol/L). |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.4. Characterization
Rheological measurements on sHG samples were carried out on a TA AR-G2 rheometer using a steel plate of 40mm diameter at 25 ℃. The frequency-sweep spectra were recorded in a constantstrain mode (ε=0.5%) over the frequency range of 0.1-100rad/s. The strain-sweep test were conducted in a constant frequency of 6.28rad/s over the strain range from 0.1 to 5000. The recovery performance of the samples in response to the applied shear forces were investigated by time-sweep and cyclic test. The sample was carefully placed between the para-plate and the platform. The programmed procedure was adopted as follows (applied shear force expressed in term of strain and duration in parentheses): 1% (400s)→1400% (100s)→1% (400s)→1400% (100s)→1% (400s)→1400% (100s)→1% (400s).
AcknowledgmentThis work was supported by the Petro China Innovation Foundation (No. 2014D-5006-0201), the National Natural Science Foundation of China (Nos. 51473152 and 51573174), and the Fundamental Research Funds for the Central Universities (Nos. WK2060200012, WK3450000001).
| [1] | T. Billiet, M. Vandenhaute, J. Schelfhout, Vlierberghe S.Van, P Dubruel. A review of trends and limitations in hydrogel-rapid prototyping for tissue engineering. Biomaterials 33 (2012) 6020–6041. DOI:10.1016/j.biomaterials.2012.04.050 |
| [2] | M. Hamidi, A. Azadi, P Rafiei. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv.Rev. 60 (2008) 1638–1649. DOI:10.1016/j.addr.2008.08.002 |
| [3] | Y. Wang, Z.H. Chai, H.Y. He, Y.Z. Jiang, G.Q Fu. Polyacrylamide hydrogels with phenylboronic acid moieties for the imprinting of proteins. Chin.Chem.Lett. 21 (2010) 1487–1489. DOI:10.1016/j.cclet.2010.06.005 |
| [4] | H.R Brown. A model of the fracture of double network gels. Macromolecules 40 (2007) 3815–3818. DOI:10.1021/ma062642y |
| [5] | S. Mora, The kinetic approach to fracture in transient networks, Soft Matter 7 (2011)4908-4917. |
| [6] | H.P. Cong, P. Wang, S.H Yu. Stretchable and self-healing graphene oxide-polymer composite hydrogels:a dual-network design. Chem.Mater. 25 (2013) 3357–3362. DOI:10.1021/cm401919c |
| [7] | D. L. Taylor, M. In Het Panhuis, Self-healing hydrogels, Adv. Mater. 28(2016) 9060-9093. |
| [8] | A. Phadke, C. Zhang, B. Arman, et al., Rapid self-healing hydrogels. Proc.Natl. Acad.Sci.U.S.A. 109 (2012) 4383–4388. DOI:10.1073/pnas.1201122109 |
| [9] | B.L. Zhu, N. Jasinski, A. Benitez, et al., Hierarchical nacre mimetics with synergistic mechanical properties by control of molecular interactions in self-healing polymers. Angew.Chem.Int.Ed. 54 (2015) 8653–8657. DOI:10.1002/anie.201502323 |
| [10] | G. Akay, ${referAuthorVo.mingEn} A.Hassan-Raeisi, D.C. Tuncaboylu, et al., Self-healing hydrogels formed in catanionic surfactant solutions. Soft Matter 9 (2013) 2254–2261. DOI:10.1039/c2sm27515e |
| [11] | X. Hao, H. Liu, Z. Lu, Y. J. Xie, H. Y. Yang, Endowing the conventional HMPAM hydrogel with pH-responsive and self-healing properties, J. Mater. Chem. A 1 (2013)6920-6927. |
| [12] | G.H. Deng, F.Y. Li, H.G. Yu, et al., Dynamic hydrogels with an environmental adaptive self-healing ability and dual responsive sol-gel transitions. ACS Macro Lett. 1 (2012) 275–279. DOI:10.1021/mz200195n |
| [13] | C. Zeng, H. Seino, J. Ren, K. Hatanaka, N Yoshie. Bio-based furan polymers with self-healing ability. Macromolecules 46 (2013) 1794–1802. DOI:10.1021/ma3023603 |
| [14] | Y.L. Zhang, B. Yang, X.Y. Zhang, et al., A magnetic self-healing hydrogel. Chem. Commun. 48 (2012) 9305–9307. DOI:10.1039/c2cc34745h |
| [15] | J. J. Cash, T. Kubo, A. P. Bapat, B. S. Sumerlin, Room-temperature self-healing polymers based on dynamic-covalent boronic esters, Macromolecules 48 (2015)2098-2106. |
| [16] | A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume, H. Yamaguchi, Macroscopic self-assembly through molecular recognition, Nat. Chem. 3 (2011)34-37. |
| [17] | Z.J. Wei, J. He, T. Liang, et al., Autonomous self-healing of poly (acrylic acid) hydrogels induced by the migration of ferric ions. Polym.Chem. 4 (2013) 4601–4605. DOI:10.1039/c3py00692a |
| [18] | B.K. Ahn, D.W. Lee, J.N. Israelachvili, J.H Waite. Surface-initiated self-healing of polymers in aqueous media. Nat.Mater. 13 (2014) 867–872. DOI:10.1038/nmat4037 |
| [19] | X.Y. Dai, Y.Y. Zhang, L.N. Gao, et al., A mechanically strong highly stable, thermoplastic, and self-healable supramolecular polymer hydrogel. Adv. Mater. 27 (2015) 3566–3571. DOI:10.1002/adma.v27.23 |
| [20] | H. Liu, C.M. Xiong, Z. Tao, et al., Zwitterionic copolymer-based and hydrogen bonding-strengthened self-healing hydrogel. RSC Adv. 5 (2015) 33083–33088. DOI:10.1039/C4RA15003A |
| [21] | D.C. Tuncaboylu, M. Sari, W. Oppermann, O Okay. Tough and self-healing hydrogels formed via hydrophobic interactions. Macromolecules 44 (2011) 4997–5005. DOI:10.1021/ma200579v |
| [22] | F. Candau, J Selb. Hydrophobically-modified polyacrylamides prepared by micellar polymerization. Adv.Colloid Interface Sci. 79 (1999) 149–172. DOI:10.1016/S0001-8686(98)00077-3 |
| [23] | E.J. Regalado, J. Selb, F Candau. Viscoelastic behavior of semidilute solutions of multisticker polymer chains. Macromolecules 32 (1999) 8580–8588. DOI:10.1021/ma990999e |
| [24] | I. Jeon, J.X. Cui, W.R.K. Illeperuma, J. Aizenberg, J.J Vlassak. Extremely stretchable and fast self-healing hydrogels. Adv.Mater. 28 (2016) 4678–4683. DOI:10.1002/adma.v28.23 |
| [25] | M.R. Caputo, J. Selb, F Candau. Effect of temperature on the viscoelastic behaviour of entangled solutions of multisticker associating polyacrylamides. Polymer 45 (2004) 231–240. DOI:10.1016/j.polymer.2003.11.010 |
| [26] | A. Hill, F. Candau, J. Selb, Properties of hydrophobically associating polyacrylamides: influence of the method of synthesis, Macromolecules 26 (1993)4521-4532. |
| [27] | X. Hao, W.F. Zhou, R.C. Yao, et al., A new class of thermo-switchable hydrogel: application to the host-guest approach. J.Mater.Chem.A 1 (2013) 14612–14617. DOI:10.1039/c3ta13250a |
| [28] | Y. L. Chen, Z. B. Guan, Multivalent hydrogen bonding block copolymers self-assemble into strong and tough self-healing materials, Chem. Commun. 50 (2014)10868-10870. |
| [29] | G.A. Williams, R. Ishige, O.R. Cromwell, et al., Mechanically robust and self-healable superlattice nanocomposites by self-assembly of single-component sticky polymer-grafted nanoparticles. Adv.Mater. 27 (2015) 3934–3941. DOI:10.1002/adma.201500927 |
| [30] | F. Chambon, H.H Winter. Linear viscoelasticity at the gel point of a crosslinking PDMS with imbalanced stoichiometry. J.Rheol. 31 (1987) 683–697. DOI:10.1122/1.549955 |
| [31] | H.H. Winter, F Chambon. Analysis of linear viscoelasticity of a crosslinking polymer at the gel point. J.Rheol. 30 (1986) 367–382. DOI:10.1122/1.549853 |
| [32] | T. Bai, S.J. Liu, F. Sun, et al., Zwitterionic fusion in hydrogels and spontaneous and time-independent self-healing under physiological conditions. Biomaterials 35 (2014) 3926–3933. DOI:10.1016/j.biomaterials.2014.01.077 |
| [33] | D.C. Tuncaboylu, M. Sahin, A. Argun, W. Oppermann, O Okay. Dynamics and large strain behavior of self-healing hydrogels with and without surfactants. Macromolecules 45 (2012) 1991–2000. DOI:10.1021/ma202672y |
| [34] | Y.L. Chen, A.M. Kushner, G.A. Williams, Z.B Guan. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat.Chem. 4 (2012) 467–472. DOI:10.1038/nchem.1314 |