 2017, Vol. 28
2017, Vol. 28 


 , Yi Liuc, Zhan-Ting Lia
, Yi Liuc, Zhan-Ting Lia
b Department of Pathology, School of Basic Medical Sciences, Fudan University, Shanghai 200032, China;
c The Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley 94720, United States
In the past several decades, great effort has been devoted to the development of methods to drug delivery and controlled release for cancer treatment [1]. Among others [2], extensively studied drug delivery systems (DDSs) include molecular and polymeric micelles and liposomes [3], dendrimers and analogues [4], hydrogels [5], carbon nanotubes and dots [6], discrete nanoparticles [7], and polymeric prodrugs [8]. In recent years, stimuliresponsive DDSs have been widely exploited due to their potential in providing tumor-specific chemotherapy [9], which is highly valuable in improving treatment efficacy and reducing systemic side effects. In this context, pH-responsive DDSs have received much attention because the difference in pH values existing between healthy tissues (ca. 7.4) and cancer tissues (ca. 4.5-6.8) can be utilized to trigger controlled release of carrier-loaded drugs in the acidic cancer cell microenvironment [10]. Efforts along this line have primarily focused on the development of DDSs that release loaded drugs in the acidic conditions of the tumor microenvironment through protonation of basic groups borne by the DDSs. Although many of these DDSs are capable of overcoming multidrug resistance (MDR) of cancer cells, which is a major obstacle for successful cancer chemotherapy, and have many other advantages, there has been no clinical success of pH-responsive DDSs in chemotherapy to date. Moreover, many of the reported DDSs depend on complicated procedures of preparation and purification. Therefore, there is a strong desire for the creation of new efficient but straightforward prototypes for DDSs.
Among FDA-approved anticancer chemical drugs, there are about 15 anionic chemical drugs that are clinically used in the form of salts with discrete counterions [11]. Most of these anionic drugs suffer from MDR of cancer cells, and several families of DDSs have been generated which involve different loading mechanisms. One straightforward, yet potentially general-purpose approach for the design of new DDSs for these anionic drugs is to develop porous (multi)cationic carriers that adsorb the drugs through electrostatic interaction, and deliver and release the drugs in cancer cells by neutralizing the drugs in the acidic microenvironment of tumors.Although the strategy of using multicationic carriers has been demonstrated to be most promising for transport of nucleic acids [12], to the best of our knowledge, this strategy has not been exploited systematically for clinically used chemotherapeutic agents.
We and other groups have recently developed the self-assembly strategy for the generation of homogeneous periodic supramolecular organic frameworks (SOFs) in water [13 -15], utilizing hydrophobically driven encapsulation of dimers of aromatic units by the cucurbit [8]uril (CB [8]) ring [16]. In particular, a three-dimensional (3D) polycationic SOF with well-defined pores was found to reversibly adsorb and release anionic organic guests in response to pH value change [13]. Herein we report that such periodic porous SOFs can be developed as drug delivery systems (DDSs). The new sof-DDSs, prepared by simply dissolving two components of defined molar ratio in water, can efficiently adsorb pemetrexed (PMX) disodium, a dianionic chemotherapeutic agent clinically used for the treatment of breast and a variety of other cancers, at physiological pH with high loading. We further demonstrate that the in situ-prepared PMX-loaded frameworks, dubbed as PMX@SOFs, can overcome MDR of human breast cancer MCF-7/ Adr cells, selectively release the drug in the acidic microenvironment of cancer cells, and kill the cancer cells with a substantially improved efficiency in both in vitro and in vivo studies.
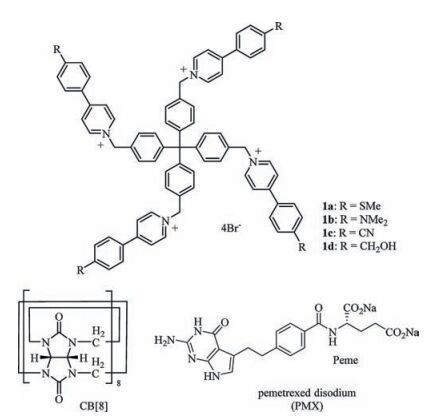
2. Results and discussionPrevious investigation showed that the 1:2 mixture of a tetracationic monomer that bears four 4-(4-methoxyphenyl)-pyridinium units and CB [8] in water produced 3D diamondoid SOF[13c]. For the present study, four new tetracationic compounds 1a-1d (Fig. 1) were prepared, which all displayed good solubility (≥2 mmol/L) in water. The compounds were introduced with different substituents at the 4-position of the peripheral benzene rings for testing their toxicity and the efficiency in transporting drugs.

|
Download:
|
| Figure 1. The structures of 1a-d, CB [8], and pemetrexed disodium. | |
{kind=link}
The binding of compounds 1a-1d with CB [8] in D2O was first studied by 1H NMR spectroscopy. The 1H NMR spectra of the solutions of their 1:2 mixtures with CB [8] (2.0 mmol/L) all displayed broad signals (Figs. S1-S4 in Supporting information), which remained unchanged at higher temperature (up to 60 ℃) or after more amount of CB [8] was added. These observations implied that stable supramolecular architectures were formed from complexation between the two compounds. The 2:1 binding stoichiometry of the 4-phenylpyridinium (PhPy) units of 1a, 1b and CB [8] was further confirmed by Job's plots using the fluorescence spectroscopy (Figs. S5-S8 in Supporting informaion). By using the competition 1H NMR method [17], we determined the apparent association constants (Ka) of the 2:1 complexes between the PhPy unit of 1a-1d and CB [8] in 50 mmol/L CD3CO2Nabuffered D2O (pD = 4.74) to be 1.4 × 10 14, 8.5 ×10 13, 1.7 × 10 14 and 8.1 ×10 13 L 2/mol2, respectively. The values are comparable to that of the first 3D SOF constructed from a methoxyl-bearing monomer[13c]. 2D 1H NMR diffusion ordered spectroscopy (DOSY) revealed nearly identical diffusion coefficients (D) — 5.5 ×10 -11, 6.03 × 10 -11, 4.22 × 10 -11, and 7.5 ×10 -11 m 2/s — for the above four 1:2 solutions ([CB [8]] = 2.0 mmol/L) in D2O (Figs. S9-S12 in Supporting information). These values were pronouncedly lower than the values (1.6 × 10 -10 -2.5 ×10 -10 m 2/s) of the corresponding four tetrahedral monomers (Figs. S9-S12 in Supporting information), supporting the formation of large supramolecular aggregates SOF-a-d (Fig. 2) from the four mixtures.

|
Download:
|
| Figure 2. Illustration of the self-assembly of 3D SOF-a from 1a and CB [8] (1:2) in water and subsequent adsorption of PMX. The space-filling structural model was obtained using Materials Studio 7.0. | |
{kind=link}
DLS experiments for the 1:2 solutions at [1] = 0.05 mmol/L gave rise to the hydrodynamic diameter (DH) of their complexes as 33, 27, 50, and 26 nm, respectively (Figs. S13-S16 in Supporting information). In contrast, the aqueous solution of the four monomers of the same concentration did not afford large aggregates (DH < 4 nm). The DH value of these 1:2 solutions further increased with the increase of the concentration and, at [1] = 2.0 mmol/L, reached 126, 98, 133, 112 nm, respectively (Figs. S17-S20 in Supporting information). Further increase of the concentration led to the formation of hydrogels or participates (for 1c). These results supported that all four mixtures formed 3D self-assembled frameworks SOF-a-d (Fig. 2), similar to the anisolederived monomer of the same backbone [13]. For all the experiments, the DH values were time-independent, indicating that the large aggregates were generated very quickly. Although the DH values of SOF-a-d were concentration-dependent, the relatively large values revealed at the lowest concentration boded well for that the framework would be able to produce important enhanced permeability and retention (EPR) effect in accumulating in cancer cells [18].
Synchrotron small angle X-scattering (SAXS) experiments were then performed for the four 1:2 aqueous solutions (CB [8] = 4.0 mmol/L). The profiles all exhibited a broad, but relatively strong peak corresponding to the d-spacing of 5.0, 4.8, 5.0, and 4.7 nm (Fig. 3a and Fig. S21a-S21c), respectively. Considering the dynamic nature of self-assembled architectures, these values matched well with the {100} spacing, which was calculated to be ~4.9 nm from the simulated crystal structure of 3D diamondoid SOF-a-d [19]. Synchrotron X-ray diffraction (XRD) studies of the same four solutions also revealed a similar broad peak—around 5.0, 4.7, 4.8 and 4.7 nm, respectively—as well as another broad peak around 1.7 nm for all the samples (Fig. 3b and Fig. S21d-S21f). The second peak was in agreement with the spacing (1.7 nm) of the {220} face calculated for the modelled 3D SOFs. The broadness of the peaks may be attributed to the dynamic nature, while structural defects in the solution-phase self-assembled frameworks may not be excluded.

|
Download:
|
| Figure 3. Solution-phase synchrotron a) SAXS and b) XRD profiles of SOF-a formed in the 1:2 solution of 1a (2.0 mmol/L) and CB [8] in water. Solid-state c) SAXS and d) XRD profiles of SOF-a obtained by evaporating the aqueous solution. The peak values were attributed by choosing the position that is highest above the straight line defined by the two saddle points of the broad peak. e) 2D synchrotron SAXS profile of solid SOF-a. f) TEM image of SOF-a. Scale bar: 1 mm. Inset: SAED pattern showing the reciprocal lattices relating to {200} and {220} facets of foursquare order. Scale bar, 2 nm. | |
{kind=link}
Evaporation of the solution of SOF-a under ambient temperature first afforded a hydrogel, which slowly solidified into microcrystals with further loss of solvent. Synchrotron SAXS profile of the microcrystals (Fig. 3c) exhibited two scattering peaks centered around 4.9 and 2.5 nm, corresponding to the {100} and {200} (2.5 nm) spacings of the modeled 3D framework. These two peaks (4.9 and 2.5 nm) were also observed on the 2D synchrotron SAXS profile (Fig. 2e). The XRD profile of the microcrystals exhibited two peaks around 4.9 and 1.7 nm (Fig. 3d), respectively, which matched the {100} and {220} spacings of the modeled 3D framework (Fig. 2). Transmission electron microscopy (HR-TEM) image revealed the formation of square microcrystals (Fig. 3f). The selected area electron diffraction (SAED) pattern (Fig. 3f, inset) clearly indicated periodicity of 2.5 and 1.7 nm that corresponds to {200} and {220} lattice spacings. All these results supported that the periodicity of the 3D framework also existed in the solid state.
The void volume of the four 3D SOFs, calculated on the basis of the modeled frameworks, was ca. 77%, whereas the pore aperture, defined by six CB [8] units in one self-assembled macrocycle adopting a cyclohexane-like chair conformation, was estimated to be about 2.1 nm. The above DLS experiments showed a size range of 26-133 nm for the four homogeneous frameworks within the investigated concentration range. This size range is very suitable for cell uptake of nanoscale drug carriers due to EPR effect [18]. We then first evaluated the in vitro cytotoxicity of SOF-a-d for multidrug resistant (MDR) human breast cancer (MCF-7/Adr) cells using the Cell Counting Kit-8 (CCK-8) assay (Fig. S22 in Supporting informaiton). It was found that the viability of the MCF-7/Adr cells maintained at about 87%, 89%, 91%, and 90% after incubating for 24 h with the SOFs at a relatively high concentration of 200 mg/mL, indicating that all the frameworks are of low cytotoxicity.
PMX disodium has been clinically used as chemotherapeutic agent for the treatment of breast cancer and a variety of other types of cancers [20]. However, PMX also suffers from MDR, due to efflux from cancer cells through different mechanisms [21], as well as inadequate delivery to target tissues and/or dose-limiting side effect [22]. Thus, the potential of homogeneous porous SOF-a-d for transporting PMX was investigated. The adsorption of PMX by SOF-a was first studied in phosphate-buffered saline (PBS) at the physiological pH of 7.4. Fluorescence titration experiments showed remarkably efficient quenching of the emission of the PhPy unit of SOF-a at very low PMX/[PhPy] molar ratio (Fig. S23 in Supporting information). In contrast, in the absence of CB [8], the quenching of the fluorescence of 1a by PMX at identical concentration was substantially lower (Fig. S23 in Supporting information). These observations revealed efficient adsorption of PMX by the selfassembled polyelectrolyte framework, which led to considerable enrichment of PMX in the interior of the framework. The fluorescence quenching was also time-independent, indicating that the adsorption of PMX into the interior of the framework took place instantaneously. The fluorescence signal of the PhPy unit of SOF-a and 1a appeared around 494 and 430 nm, respectively (Fig. S24a-d), indicating that the form signal should be generated from the excimer of the PhPy unit encapsulated in the cavity of CB [8].
We then performed dialysis experiments for all the four frameworks to further estimate their adsorption capacity for PMX (Fig. 4a). In a typical experiment, 1.0 mg of the mixture of PMX, 1a-1d and CB [8] (molar ratio: 2:1:2) was added to a dialysis bag (1 mL, cutoff Mn: 1000 Da) that was immerged in a PBS buffer (totally 10 mmol/L, 15 mL, pH 7.4) or AcOH/AcONa buffer ([AcOH] +[AcONa] = 0.1 mmol/L, 15 mL, pH 4.5). In all the mixtures, the concentration of the cationic PhPy unit of 1a-1d was equal to that of the CO2- unit of PMX. The solution was subjected to dialysis for 3 days at 37 ℃ and the buffer was renewed every 6 h. Following the adsorption of the carboxylate unit of PMX by UV-vis spectroscopy, we determined that only 12, 18, 9 and 21 mol% of PMX diffused to the buffer at pH 7.4, indicating that most of the drug was retained within the framework, which corresponded to 21, 20, 23 and 20 wt % of load capacity for the four frameworks SOF-a-d. Considering that the complexation between the frameworks and PMX was dynamic, it was expected that further elongation of the dialysis time would lead to diffusion of more PMX into the buffer. However, the very slow diffusion of the adsorbed drug to the buffer should allow enough time for PMX@SOFs to enter MDR cancer cells if the frameworks are able to exhibit important EPR effect.

|
Download:
|
| Figure 4. a) Adsorption efficiency of SOF-a-d ([PhPy] = 0.2 mmol/L) for PMX (0.1 mmol/L) determined by monitoring the absorption at -225 nm using UV-vis spectroscopy after dialysis at 37 ℃ in PBS buffer (pH 7.4) for three days. b) Release of adsorbed PMX from SOF-a-d ([PhPy] = 0.2 mmol/L) at 37 ℃ against time in PBS (pH 7.4) and MeCO2Na/ MeCO2H buffer (pH 4.5). | |
{kind=link}
Regarding the 1:1 cation/anion ratio, the above values represent remarkably higher load efficiency for the frameworks when compared with that of cationic liposomal delivery systems [23].The high load capacity of these new frameworks may be rationalized as the following: the adsorption of PMX by the homogeneous porous framework readily led to the formation of soft acid (PhPy+)-soft base (CO2-) ion pairs and hard acid (Na +)-hard base (Br-) ion pairs through ion exchange; on the other hand, the regular porosity of the homogenous frameworks minimized electrostatic repulsion of the PMX anions, which is substantial in liposomal carriers. DLS showed that after the adsorption of PMX, the DH of the frameworks was nearly unchanged (Figs. S13-S16 in Supporting information), supporting that the frameworks were maintained after adsorption of PMX. The release of adsorbed PMX from the frameworks was investigated at two pH values and the results are provided in Fig. 4b. It can be found that at normal blood plasma pH of 7.4, only very small amount (2%-4%) of the adsorbed PMX exuded after 60 h. This result supports that SOF-a-d will cause little damage to normal tissues due to minimal PMX leaching during a delivery process. At pH 4.5, a value the same as that in the lysosomal counterpart of endosomes in cancer cells[9e, 24], PMX release occurred much more quickly and about 95%, 97%, 91%, and 93% of PMX were released after 60 h. Actually, most of the drug (>75%) had been released after 20 h. This pH-responsive release profile supported that dianionic PMX should be neutralized at low pH, which led to decreased electrostatic attractions and subsequent guest release.
Internalization of SOF-a and PMX-loaded SOF-a (PMX@SOF-a) in MDR MCF-7/Adr cells was then studied by following the characteristic fluorescence associated with 1a (Fig. S24 in Supporting information). For all the following experiments, PMX@SOF-a samples were prepared in situ by dissolving the three compounds in water before use. Confocal laser scanning microscopic (CLSM) image (Fig. S25c and S25d in Supporting information) revealed that in situ-prepared SOF-a quickly entered MDR MCF-7/Adr cells after incubation. The concentration of the framework reached maximum within 30 min, as evidenced by quantitative measurement of the mean fluorescence intensity of 1a per cell via flow cytometry (Fig. S25c and S25d). Similarly, CLSM imaging showed that PMX@SOF-a could also enter MDR MCF-7/ Adr cells, but it took two hours for the fluorescence of 1a to reach maximum (Fig. S25c and S25d). This observation was consistent with the H & E and TUNEL-stained tumor tissue section experiments (vide infra, Fig. 7 and Fig. S29 in Supporting information), consistent with the release of PMX from the framework into the interior of MDR MCF37/Adr cells in response to pH change. Control experiments indicated that within the same timescale, neither 1a nor its 1:2 mixture with PMX (1:2) could enter the cancer cells at concentrations identical to that of SOF-a or PMX@SOF-a, as no significant fluorescence of 1a was detected from the cells, which may be attributed to MDR of the cells towards both samples. These results indicated that the tetrahedral monomer itself was not capable of delivering PMX to enter the cells.

|
Download:
|
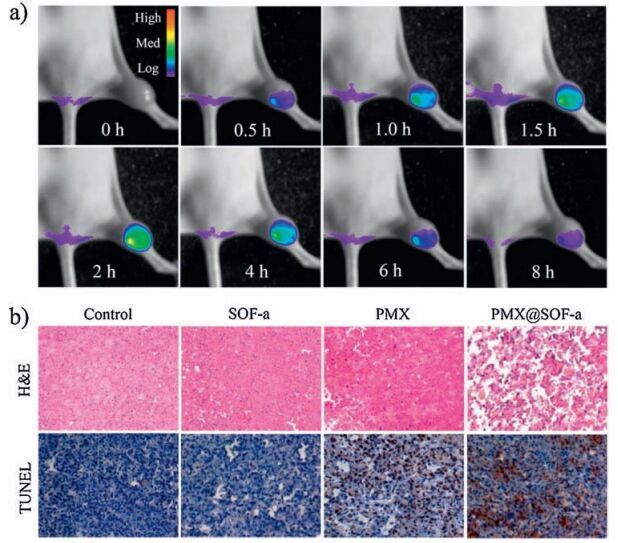
| Figure 7. a) In vivo fluorescence imaging of mouse bearing MDR MCF-7/Adr tumor on the leg after intravenous injection of PMX@SOF-a from the tail (SOF-a dosage: 50 mg/kg; PMX dosage: 10.5 mg/kg). In vivo images. b) H & E and TUNEL-stained tumor tissue sections excised from the mouse 14 days after treatment. | |
{kind=link}
It has been established that free drugs may enter MDR MCF-7/ Adr cells through a flip-flop mechanism, but could be extruded by overexpressed efflux P-glycoproteins (P-gp) [24]. We then further studied the internalization mechanism of PMX@SOF-a into MDR MCF-7/Adr cells using different endocytic inhibitors by flow cytometry (Fig. S26 in Supporting information). Compared with the internalization of PMX@SOF-a at the normal body temperature (37 ℃) in the absence of any inhibitor, the process was significantly inhibited at lowered temperature (4 ℃) or by the use of chlorpromazine (CPZ), the clathrin-mediated endocytosis inhibitor. In contrast, no inhibition was observed when the cells were pretreated with genistein (GN, caveolae-dependent endocytosis inhibitor), amiloride (AM, micropinocytosis inhibitor) or colchicine (CC, microtubule-dependent macropino-cytosis inhibitor). These results indicated that the uptake of PMX@SOF-a by the MDR cancer cells was an energy-dependent endocytosis process mediated by clathrin [25]. In this process, the frameworks were first captured by the cytosol (endosomes) after incubation with MDR MCF-7/Adr cells, and then captured by the lysosomes.
The internalization process for MDR MCF-7/Adr cancer cells was further investigated by staining the nuclei with 40, 6-diamidino-2-phenylindole (DAPI) (Fig. 5a) and lysosomes with LysoTraker Red (Fig. 5b), respectively. After incubation with PMX@SOF-a for 2 h, the cancer cells were examined by CLSM. It was found that the luminescent signals of PMX@SOF-a (Fig. 5c) and lysosome-stained LysoTraker Red were highly overlapping (Fig. 5d --f), which further confirmed that PMX@SOF-a was captured into MDR MCF-7/ADR cells by the endocytosis pathway. The delivery of fluorescence-silent PMX into the cells through the SOF carrier was confirmed by the increased activity of PMX@SOF-a as compared with that of free PMX of the identical dosage (vide infra, Figs. 6 -8).

|
Download:
|
| Figure 5. Confocal microscopic images of MDR MCF-7/Adr cells after incubation with PMX@SOF-a for 2 h ([PMX] = 0.02 mmol/L, [PhPy] = 0.2 mmol/L). a) nuclei stained DAPI; b) endosome/lysosome stained with LysoTracker Red; c) PMX@SOF-a; d) superposition of the above three images; e) bright field of image d, and f) merging of images d and e. | |
{kind=link}

|
Download:
|
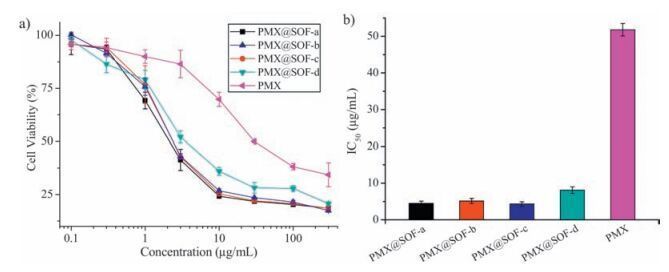
| Figure 6. a) MDR MCF-7/Adr cell viability, b) and the IC50 value of PMX@SOF-a-d and free PMX, recorded after incubation for 24 h, versus the concentration of PMX. | |
{kind=link}

|
Download:
|
| Figure 8. a) Tumor growth curves of MDR MCF-7/Adr tumors after various treatments. b) Survival rates of mice in different treatment groups within 60 days (6 mice per group, SOF-a dosage: 50 mg/kg, PMX dosage: 10.5 mg/kg, intravenous injection). | |
{kind=link}
Encouraged by the above internalization experiments, we then conducted the CCK-8 assay to investigate the ability of PMX@SOFa-d to overcome MDR of MCF-7/Adr cells (Fig. 6). It can be found that, with the identical PMX dosage, the viability of the cells treated with PMX@SOF-a-d was all considerably lower than of the cells incubated with free PMX, which clearly demonstrated that the frameworks efficiently delivered PMX to overcome MDR of the cancer cells and entered their interior to kill them. The IC50 values of PMX@SOF-a-d and free PMX were calculated to be 4.37 ± 0.65, 5.14 ± 0.71, 4.30 ± 0.63, 8.07 ± 0.91, and 51.8 ± 1.7 mg/mL, respectively, indicating that the transport of SOF-a-d caused 12, 10, 12, and 6.5-fold reduction of the dosage of PMX. Control experiments showed that the four tetrahedral molecules 1a-1d, CB [8], or the four SOF-a-d samples, with the same component concentrations as used for the above PMX@SOF-a-d, did not exhibit observable cytotoxicity for MDR MCF-7/ADR cells, while the four mixtures of 1a-d with PMX or the mixture of CB [8] with PMX, also with the same component concentrations as used for PMX@SOF-a-d, all exhibited similar, but reduced cytotoxicity for MDR MCF-7/ADR cells, with IC50 vales being comparable to that of PMX itself (Fig. S27 in Supporting information). The fact that all the four frameworks exhibited substantial promotion for the treatment efficacy of PMX suggested that the substituents at the ends of the tetrahedral monomers did not impose important influence on their delivery capacity and their porous interior should play a key role in accommodating the drug.
Flow cytometry (Fig. S28 in Supporting information) revealed that MDR MCF-7/Adr cells did not show obvious apoptosis after incubation with carriers SOF-a-d for 24 h, suggesting negligible cytotoxicity of the frameworks for the cells. For the cells which were incubated with PMX for 24 h, most of the cells were at early apoptosis state (62.5%). In contrast, most of MDR MCF-7/Adr cells incubated with PMX@SOF-a-d for 24 h were at late apoptosis state (54.6%-72.1%), which may be considered as another evidence that the frameworks raised the therapy efficacy of the adsorbed PMX.
The above cytotoxicity experiments (Fig. 6) revealed that SOF-a and SOF-c exhibited the highest transport capacity for PMX. We thus further evaluated the in vivo release and metabolism, and the therapeutic effect of PMX@SOF-a for the MDR MCF-7/Adr tumor of nude mice by monitoring the fluorescence of SOF-a. Nude mice bearing MDR MCF-7/Adr tumors were intravenously injected with the PMX@SOF-a solution from the tail, and the fluorescence intensity of the bioimaging signal of 1a at 500 nm in the tumor region was monitored (Fig. 7a). After 2 h, the fluorescence intensity reached maximum, suggesting efficient enrichment of PMX@SOF-a in the tumor region. Similar collection was also observed for PMXfree SOF-a (Fig. S29 in Supporting information), further confirming that the frameworks were capable of overcoming MDR of the cancer cells. For both experiments, the fluorescence intensity gradually weakened after 2 h, suggesting that 1a was slowly metabolized or degraded in the cancer cells. As in the first 2 h, the concentration of 1a continually increased, the framework should remain intact, even though degradation led to gradual decrease of its concentration starting after 2 h.
The treatment efficacy of PMX@SOF-a against subcutaneous MDR MCF-7/ADR tumors, in terms of apoptosis, was then evaluated in vivo. The apoptosis/necrosis of the cells, as shown by hematoxylin and eosin (H & E) and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)-staining images (Fig. 7b), were recorded after being treated with PBS, SOF-a, PMX or PMX@SOF-a. It can be found that, for the PMX@SOF-a treatment group, most tumor cells were severely destroyed. In contrast, SOF-a of the same dosage had negligible effect on the tumor cells, and PMX of the same dosage caused only small damage on the tumor cells. Tumors of the above four treatment groups (Fig. 7b) also exhibited different growth behavior (Fig. 8a).Tumors treated with PBS or SOF-a grew quickly in a comparable speed, suggesting that both the control and the framework had no inhibition capacity for the tumors. PMX alone did display observable inhibition effect. However, the size of the tumors after treatment still increased by 3 times after 14 days. In sharp contrast, PMX@SOF-a of the same dosage could completely inhibit the growth of the tumors.
The change of the body weight of tumor-bearing mice after treatment is an indicator of systemic toxicity of drug-loaded DDSs [26]. This change was simultaneously measured for the four groups of mice after they received different treatment (Fig. S30). The body weight of mice in PBS-and PMX@SOF-a-treated groups showed pronounced increase to a comparable content, while those treated with SOF-a roughly maintained their original body weight. These results suggest that both SOF-a and PMX@SOF-a did not produce severe systemic toxicity [25]. Mice treated with PMX alone displayed pronounced body weight decrease. As the above cell and tissue experiments confirmed its observable curative effect for the tumors, this body weight decrease should reasonably reflect its considerable systemic toxicity. The body weight increase for the PMX@SOF-a-treated group may be rationalized to two factors. The first is the enrichment of PMX in the tumor cells due to efficient delivery of the framework, which led to increased curative effect.The second is that much less amount of PMX circulated in the body and thus its toxicity for normal tissues could be decreased.
Survival rates of MDR MCF-7/Adr tumor-bearing mice further revealed excellent efficacy of PMX@SOF-a (Fig. 8b). Mice treated with PBS or SOF-a began to die after about 17 days and all died after about 30 days. Mice that were treated with PMX received limited curative effect. The first mouse died after 31 days, but all mice still died after 44 days. The 50% survival rate of these three groups was after 23, 24, and 38 days. In contrast, no mouse died even after 60 days for thouse treated with PMX@SOF-a. The sustained treatment of PMX@SOF-a and PMX implied that, after being released from the framework, the drug molecules could still maintain a considerable concentration, which may be attributed to its very slow degradation or metabolization in the cancer cells.
3. ConclusionWe have demonstrated for the first time the remarkable efficiency of in situ-prepared water-soluble supramolecular organic framework drug delivery systems (sof-DDSs) for highcapacity drug loading, effective drug delivery by overcoming the multidrug resistance of cancer cells, and controlled release in cancer cells by both in vitro and in vivo studies. In addition to the high drug delivery efficiency, one important advantage of the ionic sof-DDSs is that they can be obtained by simply mixing the drug and the two components of the frameworks in water, thus significantly simplifies the complicated preparation and purification processes usually required for widely investigated nanocarriers such as liposomes. Furthermore, the homogeneous nature of this new delivery system reduces aggregation-induced side effects that are sometimes induced by heterogeneous nanocarriers. The driving force for the adsorption of PMX comes from strong electrostatic interaction in aqueous media, which enables high drug load and limited release during plasma circulation and thus minimizes the side effect for normal tissues, as well as hydrophobicity. We believe that this supramolecular strategy bodes well for future development of new and efficient DDSs with appealing features that allow simple and onsite drug loading before treatment. The application of sof-DDSs for other anionic drugs of large hydrophobic segment(s) will be evaluated in the future.
4. ExperimentalAll reagents were obtained from commercial suppliers and used without further purification unless otherwise noted. All reactions were carried out under a dry nitrogen atmosphere. All solvents were dried before use following standard procedures. Column chromatography was performed on silica gel (200-300 mesh), and thin-layer chromatography (TLC) was performed on precoated silica gel plates (0.4-0.5 mm thick). Cucurbit [8]uril (CB [8]) hydrate (purity: 99+%) and 1-adamantanamine hydrochloride (purity: 99%) were purchased from J & K Scientific Co. Benzyl bromide (purity: 98%) was purchased from Sigma-Aldrich Chemical Co. Deuterium oxide (purity: 99.8 atom% D), dimethyl sulfoxide-d6 (purity: 99.5 atom% D), acetonitrile (purity: 99.5%), tetrahydrofuran (purity: >99.0%), and acetone (purity:>99.0%) were purchased from SigmaAldrich Chemical Co. Water (CHROMASOLV1 Plus, for HPLC) for preparing the samples for fluorescence, absorption, XRD, SAXS, TEM, SEM, and DLS experiments was purchased from Sigma Aldrich Chemical Co. Ultra-high-purity grade nitrogen gas (purity: 99.999%) and carbon dioxide gas (purity: 99.999%) for adsorption experiments were purchased from Shanghai Delun Gas Equipment Co.
The syntheses of compounds 1a-1d are below and others are in Supporting information.
Compound 1a: A mixture of 4-(4-(methylthio)phenyl)pyridine (0.035 g, 0.17 mmol) and tetrakis(4-(bromomethyl)phenyl)methane (0.030 g, 0.043 mmol) in THF (5 mL) and acetonitrile (10 mL) in a flask (50 mL) was stirred under reflux for 24 h and then cooled to room temperature. To the mixture was added acetone (20 mL) and the resulting precipitate was filtrated and washed with acetone and dried under vacuum. The solid was further recrystallized from acetonitrile to give compound 1a as a light yellow solid (35.4 mg, 55%). Mp >300 ℃ (decomp). 1H NMR (400 MHz, DMSO-d6) : δ 9.19 (d, 8H, J= 5.3 Hz), 8.53 (d, 8H, J= 5.4 Hz), 8.04 (d, 8H, J= 7.9 Hz), 7.56-7.39 (m, 16H), 7.21 (d, 8H, J= 7.4 Hz), 5.78 (s, 8H), 2.58 (s, 12H). 13C NMR: (100 MHz, DMSO-d6) : δ 154.29, 146.64, 145.06, 144.66, 132.45, 130.79, 128.99, 128.46, 128.18, 125.94, 123.97, 64.01, 61.48, 13.99. HRMS: Calcd. for C77H68N4S4Br2 [M]2+: 667.1402. Found: 667.1341.
Compound 1b was prepared in 31% yield as orange solid from the reaction of tetrakis(4-(bromomethyl)phenyl)methane and 4-(4-(dimethylamino)phenyl)pyridine according to a procedure as described for 1a. Mp >300 ℃ (decomp). 1H NMR (400 MHz, DMSO-d6) : δ 8.89 (d, 8H, J= 7.1 Hz), 8.33 (d, 8H, J= 7.2 Hz), 8.00 (d, 8H, J= 9.2 Hz), 7.40 (d, 8H, J= 8.4 Hz), 7.19 (d, 8H, J= 8.5 Hz), 6.86 (d, 8H, J= 9.2 Hz), 5.64 (s, 8H), 3.08 (s, 24H). 13C NMR (100 MHz, DMSO-d6) : δ 154.13, 153.10, 146.49, 143.65, 132.82, 130.75, 129.70, 127.97, 121.21, 118.54, 112.15, 63.95, 60.61, 39.60. MS (ESI): m/z 661.2
[M-2Br]2+. HRMS (ESI): Calcd. for C81H80Br2N8: 661.2431[M-2Br]2+. Found: 661.2497, HRMS (ESI): Calcd. for C81H80BrN8: 414.5224[M-3Br]3+. Found: 414.5222, HRMS (ESI): Calcd. for C81H80BrN8: 291.1621[M-4Br]4+. Found: 291.1625.
4-(Pyridin-4-yl)benzonitrile: A mixture of 4-bromobenzonitrile (364 mg, 2.0 mmol), pyridin-4-ylboronic acid (295 mg, 2.4 mmol), Pd(pddf)Cl2 (24.5 mg, 0.3 mmol), Na2CO3 (424 mg, 0.98 mmol) and 1, 2-dimethoxyethane (DME) (6 mL) and water (2 mL) in a flask (25 mL) was stirred at reflux under nitrogen for 24 h. After cooling to room temperature, the resulting mixture was diluted with 40 mL ethyl ether and then filtered. The filtrate was washed with saturated NaCl solution (30 mL). The organic phase was dried over Mg2SO4 and concentrated in vacuo. The residue was then purified by chromatography on silica gel to give 4-(pyridin-4-yl)benzonitrile as a yellow solid (295 mg, 82%). Mp: 77-78 ℃. 1H NMR (400 MHz, CDCl3) : δ 8.72 (d, 2H, J= 5.6 Hz), 7.79 (d, 2H, J= 8.3 Hz), 7.73 (d, 2H, J= 8.3 Hz), 7.50 (d, 2H, J= 5.8 Hz). 13C NMR (100 MHz, CDCl3) : δ 150.71, 146.52, 142.74, 133.03, 127.89, 121.74, 118.48, 112.99. HRMS: Calcd. for C12H8N2[M]+: 180.2053 Found: 180.2058.
Compound 1c was prepared in 53% yield as white solid from the reaction of compounds tetrakis(4-(bromomethyl)phenyl)methane and 4-(pyridin-4-yl)benzonitrile according to a procedure as described for 1a. Mp >300 ℃ (decomp). 1H NMR (400 MHz, DMSO-d6) : δ 9.36 (d, 8H, J= 6.3 Hz), 8.63 (d, 8H, J= 5.4 Hz), 8.25 (d, 8H, J= 8.2 Hz), 8.15 (d, 8H, J= 8.1 Hz), 7.49 (d, 8H, J= 8.1 Hz), 7.22 (d, 8H, J= 8.3 Hz), 5.85 (s, 8H). 13C NMR (100 MHz, DMSO-d6) : δ 153.31, 146.71, 145.17, 137.79, 133.32, 132.22, 130.77, 129.09, 128.33, 125.82, 118.07, 114.22, 64.04, 61.95. HRMS: Calcd. for C77H56N8Br2[M]2+: 625.1492. Found: 625.1494.
Compound 1d was prepared in 45% yield as yellow solid from the reaction of tetrakis(4-(bromomethyl)phenyl)methane and (4-(pyridin-4-yl)phenyl)methanol according to a procedure as described for 1a. Mp >300 ℃ (decomp). 1H NMR (400 MHz, DMSOd6) : δ 9.21 (d, 8H, J= 6.7 Hz), 8.54 (d, 8H, J= 6.2 Hz), 8.05 (d, 8H, J= 8.2 Hz), 7.57 (d, 8H, J= 7.7 Hz), 7.45 (d, 8H, J= 7.5 Hz), 7.22 (d, 8H, J= 8.0 Hz), 5.78 (s, 8H), 5.43 (s, 4H), 4.62 (s, 8H). 13C NMR (100 MHz, CDCl3) : δ 154.95, 150.19, 147.50, 146.69, 144.81, 132.44, 130.82, 128.25, 128.05, 127.31, 124.54, 64.30, 62.22, 61.59. HRMS: Calcd. for C77H68N4O4Br2[M]2+: 635.1798. Found: 635.1867.
AcknowledgmentWe thank the National Natural Science Foundation of China (Nos. 21432004, 21529201, and 91527301) and the Ministry of Science and Technology of China (No. 20 13CB834501), the Ministry of Education of China Research Fund for the Doctoral Program and of China for financial support. Shanghai Synchrotron Radiation Facility provided BL16B beamline for collecting the synchrotron X-ray scattering and diffraction data, which is also appreciated. YL thanks the support from the Molecular Foundry, Lawrence Berkeley National Laboratory, supported by the Office of Science, Office of Basic Energy Sciences, Scientific User Facilities Division, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.01.010.
| [1] | A. K. Mitra, C. H. Lee, K. Cheng(Eds. ), Advanced Drug Delivery, John Wiley & Sons, Hoboken, 2014 513 pp. |
| [2] |
(a)G. Ma, et al. , Acyclic cucurbit[n] uril molecular containers enhance the solubility and bioactivity of poorly soluble pharmaceuticals, Nature Chem. (2012); (b)D. S. Guo, K. Wang, Y. X. Wang, Y. Liu, Cholinesterase-responsive supramolecular vesicle, J. Am. Chem. Soc. 134(2012)10244-10250; (c)R. Tong, L. Tang, L. Ma, C. Tu, R. Baumgartner, J. Cheng, Smart chemistry in polymeric nanomedicine, Chem. Soc. Rev. 43(2014)6982-7012; (d)Y. Cao, X. Y. Hu, Y. Li, et al. , Multistimuli-responsive supramolecular vesicles based on water-soluble pillar[6] arene and SAINT complexation for controllable drug release, J. Am. Chem. Soc. 136(2014)10762-10769; (e)Y. X. Wang, D. S. Guo, Y. C. Duan, Y. J. Wang, Y. Liu, Amphiphilic p-sulfonatocalix[4] arene as drug chaperone for escorting anticancer, Drugs Sci. Rep. 5(2015)9019; (f)P. T. Zhang, L. Shi Huang, et al. , Self-assembled nanoparticles of amphiphilic twin drug from floxuridine and bendamustine for cancer therapy, Mol. Pharm. 12(2015)2328-2336; (g)C. Yao, P. Wang, X. Li, et al. , Near-infrared-triggered azobenzene-liposome/upconversion nanoparticle hybrid vesicles for remotely controlled drug delivery to overcome cancer multidrug resistance, Adv. Mater. 28(2016)9341-9348; (h)Y. Zhou, H. Li, Y. W. Yang, Controlled drug delivery systems based on calixarenes, Chin. Chem. Lett. 26(2015)825-828; (i)Y. Z. Chen, Y. K. Huang, Y. Chen, et al. , Novel nanoparticles composed of chitosan and b-cyclodextrin derivatives as potential insoluble drug carrier, Chin. Chem. Lett. 26(2015)909-913; (j)C. Wang, H. Zhang, D. Zeng, L. San, X. Mi, DNA nanotechnology mediated gold nanoparticle conjugates and their applications in biomedicine, Chin. J. Chem. 34(2016)299-307; (k)R. Jia, T. Wang, Q. Jiang, et al. , Self-assembled DNA nanostructures for drug delivery, Chin. J. Chem. 34(2016)265-272; (l)J. Sun, J. Wang, Z. Yang, Supramolecular assembly models of siRNA delivery systems, Chin. J. Chem. 33(2015)79-89; (m)Y. Wang, Y. Liu, Supramolecular assemblies based on p-sulfonatocalixarenes and their functions, Acta Chim. Sinica 73(2015)984-991; (n)S. Peng, J. Gao, Y. Liu, D. S. Guo, Facile fabrication of cross-linked vesicle via surface clicking of calixarene-based supra-amphiphiles, Chem. Commun. 51 (2015)16557-16560; (o)X. Wu, L. Gao, X. Y. Hu, et al. , Supramolecular drug delivery systems based on water-soluble pillar[n] arenes, Chem. Rec. 16(2016)1216-1227. |
| [3] |
(a)R. Tanbour, A. M. Martins, W. G. Pitt, G. A. Husseini, Drug delivery systems based on polymeric micelles and ultrasound: a review, Curr. Pharm. Des. 22 (2016)2796-2807; (b)T. M. Allen, P. R. Cullis, Liposomal drug delivery systems: from concept to clinical applications, Adv. Drug Deliv. Rev. 65(2013)36-48; (c)K. Wang, D. S. Guo, X. Wang, Y. Liu, Multistimuli responsive supramolecular vesicles based on the recognition of p-sulfonatocalixarene and its controllable release of doxorubicin, ACS Nano 5(2011)2880-2894; (d)L. Zhao, J. Ding, C. Xiao, et al. , Poly(L-glutamic acid)microsphere: preparation and application in oral drug controlled release, Sinica Acta Chim. 73(2015)60-65. |
| [4] |
(a)H. Wang, Q. Huang, H. Chang, J. Xiao, Y. Cheng, Stimuli-responsive dendrimers in drug delivery, Biomater. Sci. 4(2016)375-390; (b)Y. Zhou, W. Huang, J. Liu, X. Zhu, D. Yan, Self-assembly of hyperbranched polymers and its biomedical applications, Adv. Mater. 22(2010)4567-4590; (c)S. Zhang, J. Yang, M. Liu, et al. , Synthesis of peptide dendrimers and their application in the drug delivery system, Huaxue Xuebao 74(2016)401-409. |
| [5] |
(a)H. Q. Wu, C. C. Wang, Biodegradable smart nanogels: a new platform for targeting drug delivery and biomedical diagnostics, Langmuir 32(2016)6211-6225; (b)S. Liu, Y. Zhou, F. Chen, et al. , Rheological properties, drug release behavior and cytocompatibility of novel hydrogels prepared from carboxymethyl chitosan, Sinica Acta Chim. 73(2015)47-52. |
| [6] |
(a)M. Prato, K. Kostarelos, A. Bianco, Functionalized carbon nanotubes in drug design and discovery, Acc. Chem. Res. 41(2008)60-68; (b)F. Du, J. Xu, F. Zeng, S. Wu, Preparation of a multifunctional nano-carrier system based on carbon dots with pH-triggered drug release, Sinica Acta Chim. 74(2016)241-250. |
| [7] |
(a)I. Brigger, C. Dubernet, P. Couvreur, Nanoparticles in cancer therapy and diagnosis, Adv. Drug Deliv. Rev. 54(2002)631-651; (b)P. Yang, S. Gai, J. Lin, Functionalized mesoporous silica materials for controlled drug delivery, Chem. Soc. Rev. 41(2012)3679-3698; (c)T. Sun, Y. S. Zhang, B. Pang, et al. , Engineered nanoparticles for drug delivery in cancer therapy, Angew. Chem. Int. Ed. 53(2014)12320-12364; (d)Z. Tang, C. He, H. Tian, et al. , Polymeric nanostructured materials for biomedical applications, Progr. Polym. Sci. 60(2016)86-128; (e)Y. J. Chang, X. Z. Liu, Q. Zhao, et al. , P(VPBA-DMAEA)as a pH-sensitive nanovalve for mesoporous silica nanoparticles based controlled release, Chin. Chem. Lett. 26(2015)1203-1208; (f)H. Liang, H. Tian, M. Deng, X. Chen, Gold nanoparticles for cancer theranostics, Chin. J. Chem. 33(2015)1001-1010; (g)P. Yang, L. Wang, H. Wang, Smart supramolecular nanosystems for bioimaging and drug delivery, Chin. J. Chem. 33(2015)59-70; (h)X. Wang, L. Tan, Y. Yang, Controlled drug release systems based on mesoporous silica capped by gold nanoparticles, Sinica Acta Chim. 74(2016) 303-311. |
| [8] |
(a)J. Nicolas, Drug-initiated synthesis of polymer prodrugs: combining simplicity and efficacy in drug delivery, Chem. Mater. 28(2016)1591-1606; (b)Z. Du, Y. Zhang, J. Ye, H. Xu, M. Lang, Synthesis and properties of the poly (e-caprolactone)-paclitaxel prodrug, Acta Chim. Sinica 73(2015)349-356. |
| [9] |
(a)E. Fleige, M. A. Quadir, R. Haag, Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications, Adv. Drug Deliver. Rev. 64(2012)866-884; (b)S. Mura, J. Nicolas, P. Couvreur, Stimuli-responsive nanocarriers for drug delivery, Nat. Mater. 12(2013)991-1003; (c)R. Cheng, F. Meng, C. Deng, H. A. Klok, Z. Zhong, Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery, Biomaterials 34(2013)3647-3657; (d)Z. Ge, S. Liu, Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance, Chem. Soc. Rev. 42(2013)7289-7325; (e)Q. Yin, J. Shen, Z. Zhang, H. Yu, Y. Li, Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor, Adv. Drug Deliver. Rev. 65(2013)1699-1715. |
| [10] |
(a)D. Schmaljohann, Thermo-and pH-responsive polymers in drug delivery, Adv. Drug Deliv. Rev. 58(2006)1655-1670; (b)W. W. Gao, J. M. Chan, O. C. Farokhzad, pH-Responsivenanoparticles fordrug delivery, Mol. Pharm. 7(2010)1913-1920; (c)J. Liu, Y. Huang, A. Kumar, et al. , pH-Sensitive nano-systems for drug delivery in cancer therapy, Biotech Adv. 32(2014)693-710; (d)Y. J. Zhu, F. Chen, pH-Responsive drug-delivery systems, Chem. Asian J. 10 (2015)284-305. |
| [11] | https://www.cancer.gov/about-cancer/treatment/drugs. |
| [12] |
(a)D. Pissuwan, T. Niidome, M. B. Cortie, The forthcoming applications of gold nanoparticles in drug and gene delivery systems, J. Controll. Release 149(2011) 65-71; (b)X. Guo, L. Huang, Recent advancesinnonviralvectorsfor genedelivery, Acc. Chem. Res. 45(2012)971-979; (c)X. Ma, Y. Zhao, Biomedical applications of supramolecular systems based on host-guest interactions, Chem. Rev. 115(2015)7794-7839. |
| [13] |
(a)J. Tian, L. Chen, D. W. Zhang, Y. Liu, Z. T. Li, Supramolecular organic frameworks: engineering periodicity in water through host-guest chemistry, Chem. Commun. 52(2016)6351-6362; (b)K. D. Zhang, J. Tian, D. Hanifi, et al. , Toward a single-layer two-dimensional honeycomb supramolecular organic framework in water, J. Am. Chem. Soc. 135 (2013)17913-17918; (c)J. Tian, T. Y. Zhou, S. C. Zhang, et al. , Three-dimensional periodic supramolecular organic framework ion sponge in water and microcrystals, Nat. Commun. 5(2014)5574; (d)J. Tian, Z. Y. Xu, D. W. Zhang, et al. , Supramolecular metal-organic frameworks that display high homogeneous and heterogeneous photocatalytic activity for H2 production, Nat. Commun. 7(2016)11580; (e)L. Chen, Y. C. Zhang, W. K. Wang, et al. , Conjugated radical cation dimerization-driven generation of supramolecular architectures, Chin. Chem. Lett. 26(2015)811-816; (f)H. Wang, D. W. Wang, Z. T. Li, Supramolecular organic frameworks(SOFs): water-phase periodic porous self-assembled architectures, Sinica Acta Chim. 73(2015)471-479; (g)T. Wan, T. Li, From supramolecular polymers to supramolecular organic frameworks: Engineering the periodicity of solution-phase self-assembled architectures, Photochem. Imag. Sci. 33(2015)3-14; (h)L. Zhang, Y. Jia, H. Wang, et al. , pH-Responsive single-layer honeycomb supramolecular organic frameworks that exhibit antimicrobial activity, Polym. Chem. 7(2016)1861-1865; (i)L. Zhang, T. Y. Zhou, J. Tian, et al. , A two-dimensional single-layer supramolecular organic framework that is driven by viologen radical cation dimerization and further promoted by cucurbit[8] uril, Polym. Chem. 5(2014) 4715-4721. |
| [14] | M. Pfeffermann, R. Dong, R. Graf, Free-standing monolayer two-dimensional supramolecular organic framework with good internal order. J. Am.Chem.Soc. 137 (2015) 14525–14532. DOI:10.1021/jacs.5b09638 |
| [15] |
(a)Y. Zhang, T. G. Zhan, T. Y. Zhou, et al. , Fluorescence enhancement through the formation of a single-layer two-dimensional supramolecular organic framework and its application in highly selective recognition of picric acid, Chem. Commun. 52(2016)7588-7591; (b)S. Q. Xu, X. Zhang, C. B. Nie, et al. , The construction of a two-dimensional supramolecular organic framework with parallelogram pores and stepwise fluorescence enhancement, Chem. Commun. 51(2015)16417-16420. |
| [16] |
(a)Y. H. Ko, E. Kim, I. Hwang, K. Kim, Supramolecular assemblies built with host-stabilized charge-transfer interactions, Chem. Commun. (2007)1305-1315; (b)Z. J. Zhang, Y. M. Zhang, Y. Liu, Controlled molecular self-assembly behaviors between cucurbituril and bispyridinium derivatives, J. Org. Chem. 76(2011) 4682-4685; (c)Y. Liu, H. Yang, Z. Wang, X. Zhang, Controlled molecular self-assembly behaviors between cucurbituril and bispyridinium derivatives, Chem. Asian J. 8(2013)1626-1632; (d)J. Liu, C. S. Y. Tan, Y. Lan, O. A. Scherman, Aqueous polymer self-assembly based on cucurbit[n] uril-mediated host-guest interactions, Macromol. Chem. Phys. 217(2016)319-332; (e)J. Tian, L. Zhang, H. Wang, D. W. Zhang, Z. T. Li, Supramolecular polymers and networks driven by cucurbit[8] uril-guest pair encapsulation in water, Supramol. Chem. 28(2016)769-783. |
| [17] | S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P.Y. Zavalij, L Isaacs, The cucurbit[n] uril family:prime components for self-sorting systems. J.Am. Chem.Soc. 127 (2005) 15959–15967. DOI:10.1021/ja055013x |
| [18] |
(a)J. Fang, H. Nakamura, H. Maeda, The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect, Adv. Drug Deliv. Rev. 63(2011)136-151; (b)Y. Barenholz, Doxil1, The first FDA-approved nano-drug: Lessons learned, J. Control. Release 160(2012)117-134. |
| [19] | Accelrys Materials Studio Release Notes, Release 5. 0, Accelrys Software Inc, San Diego, 2008. |
| [20] |
(a)M. H. Cohen, R. Justice, R. Pazdur, Approval summary: pemetrexed in the initial treatment of advanced/metastatic non-small cell lung cancer, Oncologist 14(2009)930-935; (b)M. Hazarika, R. M. White, J. R. Johnson, R. Pazdur, FDA drug approval summaries: pemetrexed(Alimta®), Oncologist 9(2004)482-488. |
| [21] | M. Vandana, S.K Sahoo, Reduced folate carrier independent internalization of PEGylated pemetrexed:a potential nanomedicinal approach for breast cancer therapy. Mol.Pharm. 9 (2012) 2828–2843. DOI:10.1021/mp300131t |
| [22] | Y. Pluen, Role of tumor-host interactions in interstitial diffusion of macromolecules:cranial vs.subcutaneous tumors. Proc.Natl.Acad.Sci.U.S.A. 98 (2001) 4628–4633. DOI:10.1073/pnas.081626898 |
| [23] | E.N. Eldin, H.M. Elnahas, M.A.E. Mahdy, T Ishida, Liposomal pemetrexed: formulation, characterization and in vitro cytotoxicity studies for effective management of malignant pleural mesothelioma. Biol.Pharm.Bull. 38 (2015) 461–469. DOI:10.1248/bpb.b14-00769 |
| [24] | J. Han, K Burgess, Fluorescent indicators for intracellular pH. Chem.Rev. 110 (2010) 2709–2728. DOI:10.1021/cr900249z |
| [25] | Y. Tian, X. Jiang, X. Chen, Z. Shao, W Yang, Doxorubicin-loaded magnetic silk fibroin nanoparticles for targeted therapy of multidrug-resistant cancer. Adv. Mater. 26 (2014) 7393–7398. DOI:10.1002/adma.v26.43 |
| [26] | S. Tang, Q. Yin, Z. Zhang, Co-delivery of doxorubicin and RNA using pH-sensitive poly(b-amino ester)nanoparticles for reversal of multidrug resistance of breast cancer. Biomaterials 35 (2014) 6047–6059. DOI:10.1016/j.biomaterials.2014.04.025 |