 2017, Vol. 28
2017, Vol. 28 



Recently, the molecular-ionic complexes of M (Ⅱ) halides (where M=Zn) with organic amines have become a growing research interest [1-3], zinc coordination chemistry, involving a variety of nitrogen ligands, is of fundamental importance in chemistry, due to not only their intriguing architectures and various topologies [4-6], but also due to fundamental importance in chemistry, biology and materials science [7], their potential applications in areas and medicine. Such materials are frequently characterized by their various physical and chemical properties, like magnetic or ferroelectric transitions, conductivity (superconductivity), electroluminescence and photoluminescence [8-19].
Zinc chloride has become larger focus of interest in such applications, such it is one of a small number of metal halides which can be easily vitrified without any other additive components, and is therefore considered as a key compound in the research of halide glasses. Also considerable progress has been achieved in the synthesis of organic-inorganic hybrid due to their compositional diversity and rich structural chemistry [20, 21]. Therein, open-framework zinc chloride occupy an apical position and a large number of such solids with zero-, one-, two-, and three-dimensional structures have been prepared and characterized with diffraction and spectroscopic measurements.
1, 2-Diaminocyclohexane (1, 2-C6H10(NH2)2) has been widely used as organic ligands for the construction of inorganic-organic hybrid materials, which are of great interest in recent years [22-25]. Few papers with the assignments of internal vibrations of 1, 2-C6H10(NH2)2 molecule have already been published. However, numerous structural and vibrational studies on zinc derivatives have been published. The specific acidity of particular derivative allows us to choose the appropriate base in the crystal engineering of new materials.
Considering the attractive properties and attributes of halogenozincate (Ⅱ) and the new promising opportunities they may open with regard to the development of useful inorganic-organic hybrid materials. The aim of this work is to investigate a novel approach for the preparation and discussion the results of a structural investigation pertaining to a new solid solution: [1, 2-C6H10(NH3)2]ZnCl4. We have performed X-ray diffraction measurements which provide us with information about the complete crystal structure of the new compound at room temperature. In addition, we present a Hirshfeld surface analysis of the crystal structures using Crystal Explorer experimental and theoretical analysis of a new compound. This research work elucidates the structural, vibrational (FT-IR and Raman) and dielectric properties of a new zero dimensional compound [1, 2-C6H10(NH3)2]ZnCl4.
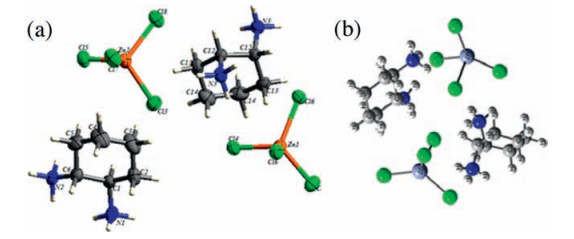
2. Results and discussion 2.1. Crystallographic studyThe title compound [1, 2-C6H10(NH3)2]ZnCl4 crystallizes in the monoclinic space group C2/c. Correspondingly, there are twelve formula units per unit cell. The crystallographic analysis of this compound reveals that the asymmetric unit is formed by two unequivalent 1, 2-diammoniumcyclohexane cations and two types of chloro-zincate tetrahedra. A view of the formula unit of the structure drawing with 50% probability thermal ellipsoids is depicted in Fig. 1a. Four amino groups of the cations ring are protonated. Thus, to ensure charge equilibrium, the organic molecule exhibited a regular configuration with normal C-C and C-N distances and C-C-C and C-C-N angles. While the C-C bond lengths varied from 1.503(6) Å to 1.523(6) Å, the main value of the C-N length of the [1, 2-C6H10(NH3)2]2+ varied from 1.492(5) Å to 1.508 Å (Table 1). Those values are comparable to the corresponding values previously reported for similar complexes [26]. In the structural arrangement, two types of zinc (Ⅱ) atom are present; Zn1 atom occupies the special positions while the Zn2 atom occupies the general positions and each atom surrounded by four chlorine atoms.

|
Download:
|
| Figure 1. Atom numbering scheme for the title compound (a: experimental; b: B3LYP/LanL2DZ optimized geometries counterpart). | |
|
|
Table 1 Comparison between observed and calculated bond length (Å) and bond angles (°) of the title compound. |
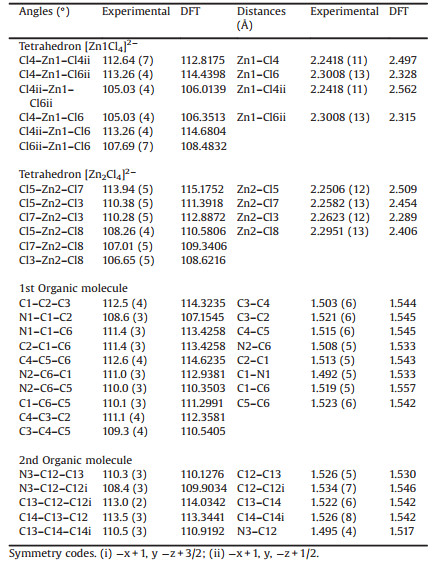
Generally, the Zn-Cl bond lengths and Cl-Zn-Cl bond angles in the [ZnCl4]2- anion are not equal to one another, but rather vary with the environment around the Cl atoms [27]. In the title compound, the four chlorine atoms of the [ZnCl4]2- anion are acting as acceptors of the hydrogen bonds. Taking into account the bond lengths and angles within the organic ions and considering the calculated average values of the Baur distortion indices [28], {ID Zn (1)-Cl=0.003(4), ID Cl-Zn (1)-Cl=0.013(2); ID Zn (2)-Cl=0.007(2), ID Cl-Zn (2)-Cl=0.0112(3)}, we deduce that all [ZnCl4]2-tetrahedra are slightly distorted, yet the [Zn (1) Cl4]2- tetrahedron is considerably distorted. The bond lengths and angles are listed in Table 1.
The organic cations are linked to the anionic unit through relatively weak N-H…Cl hydrogen bonds. They are partly responsible for the distorted tetrahedral coordination-differences between equivalent Zn-Cl bond lengths as well as Cl-Zn-Cl angles. The presence of the hydrogen bonds leads to the elongation of the terminal Zn-Cl bonds in comparison with the remaining distances between the Zn and Cl atoms. Fig. 2a shows that the atomic arrangement of the title hybrid material can be described as an alternation of organic and inorganic layers stacked. This Fig. 2b shows also the different alternation of the two organic molecules along c-axis. The tetrachlorozincate anions are located between cations and connected to the halogen atoms by weak N-H…Cl hydrogen bonds (Fig. S1 in Supporting information). All the hydrogen bonds give rise to a three-dimensional network in the structure and add stability to this compound. The parameters of those hydrogen bonds are shown in Table S3 in Supporting information. As can be noticed from Table 1, most of the computed bond lengths are slightly longer the experimental one. These discrepancies can be explained by the fact that the calculations assume an isolated molecule where the intermolecular Coulombic interaction with the neighboring molecules is absent, whereas the experimental result corresponds to interacting molecules in the crystal lattice.

|
Download:
|
| Figure 2. Projection of the atomic arrangement of [1, 2-C6H10(NH3)2]ZnCl4 structure (a: along the b axis; b: the view of the crystal packing). | |
2.2. Hirshfeld surface analysis
Molecular Hirshfeld surfaces (HS) [29] in the crystal structure have been constructed based on the electron distribution calculated as the sum of spherical atom electron densities [30]. The HS analysis is a novel and unique tool of visualizing the intermolecular interactions; it can be compared to the van der Waals envelope which other molecules or atoms come into contact with when interactions are present.
We assessed the performance of Hirshfeld surfaces analysis using several descriptors such as (curvedness of the surface) [31] and (dnorm) [32]. The contribution of each descriptor to the HS method allows us to gain an insight into these interactions.
For each point on that isosurface two distances are defined: de, the distance from the point to the nearest nucleus external to the surface, and di the distance to the nearest nucleus internal to the surface. The normalized contact distance (dnorm) based on both de and di, and the vdW radii of the atom, given by Eq. (1), which enables identification of the regions of particular importance to intermolecular interactions [33].

|
(1) |
The HS and 2-D fingerprint plots of the title compound were prepared by CrystalExplorer [34], which accepts a structure input file in the experimental CIF (Crystallographic Information File) format.
The 2-D fingerprint plot (FP) format is generated by the combination of d e and di. The 3-D HS and corresponding 2-D fingerprints were prepared for the structure using the program CrystalExplorer. In terms of an analysis of the intermolecular approaches, we find the entire interaction network fully described by the analysis of intra-and inter-molecular-cation approaches.
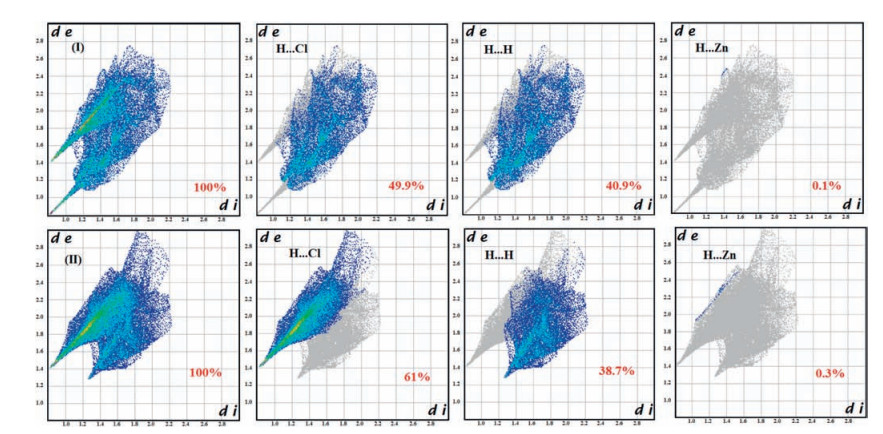
HS analysis is convenient way to get an overall picture of close intermolecular interactions and weak contacts (H…H, H…Cl, H…Zn) identifying differences in the packing motifs. The information regarding intermolecular interactions which are presented in Figs. S2a and S2b in Supporting information are visible by the spots on the Hirshfeld surfaces. The large circular depressions on the (dnorm) surface are due to the N-H…Cl bonding contacts, whereas other visible spots are due to H…H contacts. The small extent of area and light color on the surfaces are the indicator of weaker contact which are not classical hydrogen bond. By using the breakdown of fingerprint plots (existing techniques and tools based on the Hirshfeld surface and already incorporated in Crystal Explorer computer program) we can decompose fingerprint plots of organic components to highlight particular close contacts. This decomposition enables to separate contributions from different interaction types, which commonly overlap in the full fingerprint. Fig. 3 illustrates this decomposition of the fingerprint plots for two organic components in crystal lattice, highlighting separately the H…Cl, H…H intermolecular contacts and H…Zn. To provide context, the outline of the full fingerprint is shown in grey and the separate contact area in blue. In this context, the Hirshfeld surfaces of the title complex are illustrated in Figs. S2, showing surfaces that have been mapped over dnorm, de, Shape-Index and curvedness. Fig. S3 in Supporting information shows the relative contributions to the Hirshfeld surface area coming from H…Cl, H…H and H…Zn. From this analysis, it immediately emerges that 1, 2-diammoniumcyclohexane species are obviously contributor in H…Cl interactions (49.9% area for organic moiety (Ⅰ), 61% area for organic moiety (Ⅱ)). It is clear that the nature of the H…Cl contacts in two organic molecules is strikingly similar to each other. H…H contacts in organic components (Ⅰ) and (Ⅱ) are comparable, 49.9% area for organic moiety (Ⅰ) and 38.7% area for organic moiety (Ⅱ). Fig. S3 also reveals that organic moiety (A) have weak H…Zn intermolecular contacts in crystal lattice (0.1% H…Zn area for organic moiety, 0.3% H…Zn area for organic moiety (Ⅱ)). It can be concluded that the hydrogen bond interaction plays a crucial role in the construction of the 3-D architecture, especially the H…H interaction which overruns the classic hydrogen bond interactions; van der Waals force between the peripheral atoms of component units cannot be ignored.

|
Download:
|
| Figure 3. Fingerprint plots for organic moieties (Ⅰ) and (Ⅱ); showing reciprocal contacts and resolved into: H…Cl, Hglyph5H, and H…Zn. The full fingerprint appears beneath each decomposed plots as a grey shadow. | |
2.3. NMR spectroscopy
The 13C NMR spectrum of 1, 2-diammoniumcyclohexane tetrachlorozincate (Ⅱ), recorded between -10 and 70 ppm, is presented in Fig. S4 in Supporting information. The deconvolution of the experimental 13C NMR spectrum (blue) using Lorentzian and Gaussian Functions was performed with the Dmfit software [35]. The results of the deconvolution peaks are shown with different colors, leading to the presence of seven peaks corresponding to twelve carbon atoms in non-equivalent positions. This shows the existence of two organic cations in the asymmetric unit of the compound. The red spectrum confirms the good result of the deconvolution by superimposition with the experimental result (blue spectrum). This result confirms the crystallographic study, thus proving that this crystal is pure. The chemical shift to a higher frequency, 54.44, 53.79 and 49.53 ppm, can be explained by the fact that the four carbon atoms C (1), C (2), C (12) and C (12i) of the cyclic ring are linked to the electronegative atoms N1, N2 and N3 [36]. The isotropic shift at about 32.16, 26.91 and 24.62 ppm can be assigned to the carbon atoms pairs respectively away from any interaction: C5, C2; C13, C13i and C3, C4, away from any interaction. The peaks of two carbon atoms C (14) and C (14i) are observed at around 18.72 ppm (Table S4 in Supporting informa tion).
2.4. Vibrational studiesIR and Raman spectra of [1, 2-C6H10(NH3)2]ZnCl4 crystal are performed and evaluated on the basis of the characteristic vibrations of chlorozincate anion, amine group and methylene group. The molecular structure of 1, 2-diammoniumcyclohexane tetrachlorozincate (Ⅱ) with atom numbering scheme adopted in the computations is given in Fig. 1b.
The calculated vibrational wavenumber and the atomic displacement corresponding to the different normal modes are used for explicitly identifying the vibrational modes. The predicted infrared intensities and Raman activities corresponding to various normal modes were are reported in Table S5 in Supporting information along with detailed assignments. The crystal structure of the title compound consists of isolated [ZnCl4]2- tetrahedra surrounded by organic cations. Then, in order to take into account the effect of the intermolecular N-H…Cl hydrogen bonds on geometrical parameters and vibrational spectroscopy, the [ZnCl4]2- tetrahedra surrounded by two protonated 1, 2-diammonuimcyclohxane cations is perfect to model the system. For visual comparison, the observed and simulated FT-IR and Raman spectra are presented in Figs. 4 and 5. On the basis of our DFTcomputations as a primary source of assignment and on the basis of the previously reported vibrations study of a similar compound, we have assigned all vibrational bands. The frequencies of the calculated and observed bands are provided in Table S5 in Supporting information. The correlation graphs are shown in Figs. S5a and b in Supporting information, respectively for FT-IR and Raman frequencies.

|
Download:
|
| Figure 4. Experimental (black) and theoretical DFT/B3LYP/LanL2DZ (blue) FT-IRspectra of [1, 2-C6H10(NH3)2]ZnCl4. Compound at room temperature. | |

|
Download:
|
| Figure 5. Experimental (black) and theoretical DFT/B3LYP/LanL2DZ (blue) Ramanspectra of [1, 2-C6H10(NH3)2]ZnCl4. Compound at room temperature. | |
2.4.1. Internal modes
We now discuss the internal modes of the organic cation observed in the FT-IR and Raman spectra between 400 and 4000cm-1 frequency range. The experimental band frequencies are compared with those predicted by DFT calculation.
2.4.2. External modesThe Raman lines observed between 50cm-1 and 400cm-1 are assigned to the translational and vibrational modes of the organic and inorganic groups in the unit cell. The asymmetric and symmetric stretching modes of Zn-Cl appeared at 278 and 191cm-1, respectively. The DFT calculations yielded those modes at 301 and 196cm-1 [37]. Fig. 5 shows the experimental and calculated Raman spectra, respectively. It can be seen that the agreement between the observed and calculated frequencies is reasonable, showing that the DFT/B3LYP/LanL2DZ performed for a simple system built of two 1, 2-diammonuimcyclohxane cations and [ZnCl4]2- anion has accurately modeled the system.
2.4.3. Vibration of NH3+ groupWe have assigned all these bands to N-H stretching mode. As the N-H group of the (NH3+) is involved in hydrogen bonds, the wavenumbers of this mode is shifted towards lower values. In fact, the XRD study of the title compound reveals that each one of the three hydrogen atoms in (NH3+) moiety is involved in N-H…Cl hydrogen bonds with the lengths of the order of 3.192-3.427Å [38].
Thus, it can be expected that the various stretching N-H modes from 3400cm-1 to 3000cm-1 depending on the strength of hydrogen bonds. For saturated amines, it is established that the asymmetric NH3 stretch will give rise to a band between 3000cm-1 and 3500cm-1. The Raman spectrum shows a weak band observed at 3240cm-1 corresponding to NH3 asymmetric stretching mode. The symmetric stretching mode is also observed as weak intense shoulder in Raman spectrum at 3091cm-1. The DFT computations give the frequency of these bands at 3246cm-1 for NH3 asymmetric stretch and 3039cm-1 for the symmetric stretch. The observed NH3 stretching frequencies are higher than the computed frequencies due to the steric interactions (depends upon the substituents are in the axial or equatorial) [39].
The shoulder IR band observed at 1606cm-1 is assigned to the asymmetric δ(NH3) bending mode. The corresponding theoretically predicted value is at 1616cm-1. The strong band at 1502cm-1 in the IR spectrum is assigned to the NH3+ scissoring mode. The deformation, rocking and the torsional modes related to NH3+ are observed and assigned.
2.4.4. Vibration of CH2 groupThe CH2 asymmetric stretching vibrations are generally observed in the 3000-2900cm-1 region, while the CH2 symmetric stretch will appear between 2900 and 2800cm-1 [40, 41]. In our case, the CH2 asymmetric and symmetric stretching vibrations are observed in the IR spectrum at 2942 and 2872cm-1, respectively. The DFT calculation gives νas(CH2) at 3008, 3037 and 2979cm-1, whereas the symmetric band is probably masked by the band at 2461cm-1. The (CH2) deformation mode is observed as a strong band at 1447cm-1 in Raman spectrum and at 1445cm-1 as weak band in IR spectrum. Our DFT calculations predict this mode at 1470 and 1466cm-1. The band observed in the Raman spectrum at 1321cm-1 is assigned tothe (CH2) bending mode. The CH2 wagging mode appears at 1361cm-1 in the Raman spectrum and it is predicted theoretically at 1370cm-1. The peaks observed at 1266 and 1255 cm-1 in the Raman spectrum are assigned to CH2 twisting vibrations. The CH2 rocking vibrations are also identified and assigned. The detailed assignment of all observed bands related to CH2 is given in Table S5. It is interesting to note that the vibrational mode CH2 groups do not deviate much from their expected values, suggesting that the interaction of these groups with environment is not strong.
2.4.5. Vibrations of C-N, C-C, C-N-C and C-C-N groupsThe weak Raman band observed at 1032 cm-1 and the corresponding IR band at 1024 cm-1 can be assigned to the C-NH3 stretching mode. This mode is well reproduced by DFT calculations at 1015 cm-1. The medium located at 1022 cm-1 in Raman spectrum and the shoulder observed at 986 cm-1 in IR spectrum are assigned to the C-C stretching mode. The weak intensity band observed at 900 cm-1 in Raman spectrum and the very shoulder one at 978 cm-1 in IR spectrum is assigned to C-C stretching mode coupled with the rocking mode of (NH3+). The C-C-N asymmetric stretching modes are observed at 928 cm-1 in IR spectrum and at 989 cm-1 in Raman spectrum. The C-C-N deformation mode appears as a very shoulder band at 491 cm-1 in IR spectrum and at 495 cm-1 in Raman spectrum. A detailed assignment of the skeletal vibration is given in Table S5.
2.5. Thermal analysisFig. 6 display the results of the thermal analysis, the DSC curve for [1, 2-C6H10(NH3)2]ZnCl4 crystal, recorded on heating between 200 K and 550 K with a scanning rate of 5 K/min. Two endothermic peaks are existed in the DSC thermogram, which are located at 315 K and 425 K. The first is attributed to a phase transition and the second to a fusion. The TGA analysis confirm this results, no weight loss was detected between room temperature and 425 K.

|
Download:
|
| Figure 6. TGA and DSC curves of [1, 2-C6H10(NH3)2]ZnCl4. | |
2.6. Dielectric studies
The complex permittivity formalism has been employed to reveal significant information about the chemical and physical behavior of the electrical and dielectric properties, it is expressed as [42, 43].

|
where ε' and ε" are the real and imaginary parts of the dielectric constant, respectively. The variation of ε' and ε" with frequency and temperature are depicted in Figs. 5 and 6.
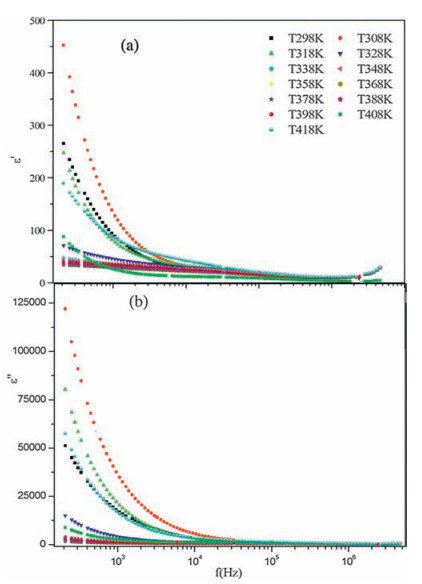
2.6.1. Frequency dependence of dielectrical studiesThe dielectric relaxation is described by a non-Debye model which gives the frequency dependent complex permittivity in the form [44]. The dielectric constant ε' decreases at low frequencies and remains constant at higher frequencies. In composites the higher value of dielectric constant at low frequencies can be associated with heterogeneous conduction [45]. The polaron hopping mechanism can also result in electronic polarization contributing to low frequency dispersion (Fig. 7a) [46]. Fig. 7b shows the variation of ε" with frequency at various temperatures. It is observed also that ε", decreases with increasing frequency for all temperatures. At low frequencies, as the temperature increases, ε" shows a dispersive behavior, while it merges at frequency above 105 Hz. The higher values of ε" at low frequencies suggest the existence of electrode polarization [47, 48].

|
Download:
|
| Figure 7. (a) Real and (b) imaginary part of part of dielectric constant as a function offrequency at several temperatures for the [1, 2-C6H10(NH3)2]ZnCl4 compound. | |
2.6.2. Temperature dependence of dielectrical study
The temperature dependence of the real (ε') and imaginary (ε") parts of the dielectric permittivity of [1, 2-C6H10(NH3)2]ZnCl4 compound measured at selected frequencies 209 Hz, 253 Hz, 306 Hz, 492 Hz, 721 Hz and 1700 Hz which are displayed in Fig. 8a and b. This evaluation was performed in the 298 K-418 K temperature range. We note the presence of a prominent dielectric peak at T=314 K, which suggests the presence of a ferroelectric transition observed in the DSC curve. This transition may be due to the superposition of many mechanisms, such as the reorientation of the ammonium groups and the break of hydrogen bonds. The real (ε') and imaginary (ε") permittivity around T=314 K increase considerably as frequency decreases. This is in agreement with the large conductivity present in this material [49].

|
Download:
|
| Figure 8. (a) Real and (b) imaginary part of dielectric constant as a function offrequency at several temperatures for the [1, 2-C6H10(NH3)2]ZnCl4 compound. | |
3. Conclusion
A novel inorganic-organic hybrid compound, 1, 2-diammonium cyclohexane tetrachlorozincate (Ⅱ) has been successfully synthesized at room temperature by slow evaporation. This compound belongs to the monoclinic system with C2/c space group. The structure of this compound consists of alternating organic and inorganic layers. The organic cations are linked to the inorganic layers by N-H…Cl hydrogen bonds. Hirshfeld surface fingerprint plots show different types of intermolecular interactions including hydrogen bonding. Besides, the geometric parameters and vibrational frequencies of [1, 2-C6H10(NH3)2]ZnCl4 have been investigated by DFT/B3LYP/LanL2DZ method. All the experimental vibrational bands have been discussed and assigned to normal mode or to combinations on the basis of our calculations. The thermal and dielectric measurements prove that the compound presents a ferroelectric phase transition.
4. Experimental 4.1. Synthesis of [1, 2-C6H10(NH3)2]ZnCl4compoundColorless crystals of [1, 2-C6H10(NH3)2]ZnCl4 were recovered in two stages: Firstly, a batch of seed material was prepared from an aqueous solution containing 38% HCl (7 mL) to (0.25 mL) of 1, 2-diaminocyclohexane. Secondly, stoichiometric quantities of ([1, 2-C6H10(NH3)2]2+, 2Cl-) and zinc chloride (ZnCl2) (molar ratio 1:1) were dissolved. After several days of slow evaporation of the solution at room temperature, colorless single crystals suitable for X-ray structure determination were obtained from the mother liquor.
4.2. ApparatusThe FT-IR spectra were obtained in the 400-4000 cm-1 region using a Nicole impact 410 FT-IR spectrophotometer with a sample depressed in a KBr pellet. The pellets were prepared by mixing 15 mg of a powder sample with 300 mg of KBr (The KBr was dried at 383 K) and compressing the whole into a disk.
Raman scattering investigations were performed using as excitation the 514.5 nm radiation of an Ar-Kr laser, and a T64000 multichannel Raman spectrometer (Horiba-Jobin-Yvon) equipped with a cooled CCD detector. The laser power in the sample was limited to 4 mW. The spectrum was measured from 50 cm-1 to 4000 cm-1.
The CP/MAS-NMR experiments were performed at room temperature on a Bruker WB 300. Then, the powdered sample was packed in a 4 mm diameter rotor and set to rotate at a speed up to 11 kHz in a Doty MAS probehead. The 13C spectrum was collected by a cross-polarization of proton with 5 ms contact time. All chemical shifts (δ) are given with respect to tetramethylsilane for 13C. Spectrum simulation was performed by using DMFIT software [35].
Thermogravimetric analysis (TGA) was carried out using an ATG PYRIS 6 instrument at the temperature range from 298 K to 450 K with a ramp rate of 5 K/min under a nitrogen atmosphere.
The differential scanning calorimetry analysis is performed using a (SETARAM DSC 131-ks) instrument in the temperatures range 290 K-450 K at the heating rate of 5 K min-1, with a polycrystalline sample is placed in a hermetic aluminum cell in a nitrogen atmosphere.
4.3. CalculationsDensity functional theory (DFT) has been treated extensively in theoretical chemistry to study of chemical systems in the last 30 years [50]. It has performed to investigate and describe the various properties of molecules, such as geometries, energies, reaction mechanisms, and spectroscopic properties.
The molecular geometry and vibrational wavenumbers were also calculated using this method using the Gaussian 03 program [51]. It is to be noted that the LanL2DZ [52] was used as basis for all atoms. Therefore, the calculations converged to optimized geometries which corresponded to true energy minima, as revealed by the lack of imaginary values in the vibrational mode calculations. Besides, the vibrational frequencies calculations were performed at the DFT/LanL2DZ level [53]. Vibration mode assignments were made, on one hand, by comparison with previous works on analogous compounds.
Before comparing the calculated vibrational frequencies with their experimental counterparts, the former were scaled by 0.9679 for the B3LYP method. The Gauss view program [54] was employed to generate visual presentations and verify the normal mode assignments.
4.4. X-ray data collectionAll reagents were purchased commercially. The X-ray data collection was carried out on a Bruker APEXⅡ CCD four circle diffractometer using MoKa radiation. The complex was collected at 296(2) K. Intensity data were collected in the ω-2θ scan mode using graphite. Automated search for space group available in WINGX was found [55] Zinc and chlorine atoms were located using the Patterson method with the program SHELXS 97 program [56]. The organic moieties were found from successive difference Fourier calculations using SHELXL 97 [57]. All the hydrogen positions of the diprotonated cation were placed geometrically and held in the riding mode (the C-H and N-H bonds were fixed at 0.97 and 0.89 Å, respectively). Full details of the refinement can be found in the supplementary materials. The refinement was performed by full-matrix least squares methods (SHELXL 97 program) and converged to an acceptable final agreement factor. Numerical details for the data collection and structure refinement of the title complex are given in Table 2. The structural graphics of the asymmetric unit was founded by ORTEP [58] (Fig. 1a) and for the other figures with the DIAMOND program [59].
|
|
Table 2 Summary of crystal data, intensity measurements and refinement parameters for[1, 2-C6H10(NH3)2]ZnCl4 crystal. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors would like to thank the members of the unit of common services, in particular Mr. Tarek Gargouri, at the University of Sfax for their assistance for the measurements of X-ray diffraction. They are also thankful to Prof Hamadi Khemakhem for this co-operating in the Raman spectroscopy measurement.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.10.002.
| [1] | M. Ben Gzaiel, A. Oueslati, J. Lhoste, M. Gargouri, A. Bulou, Synthesis, crystal structure and high temperature phase transition in the new organic-inorganic hybrid[N (C4H9)4]3Zn2Cl7H2O crystals. J. Mol. Struct. 1089 (2015) 153–160. DOI:10.1016/j.molstruc.2015.01.040 |
| [2] | J.A. Zhao, Y. Guo, J.Y. Hu, Potential anticancer activity of benzimidazolebased mono/dinuclear Zn (Ⅱ) complexes towards human carcinoma cells. Polyhedron 102 (2015) 163–172. DOI:10.1016/j.poly.2015.09.057 |
| [3] | N. Chihaoui, B. Hamdi, T. Dammak, R. Zouari, Molecular structure, experimental and theoretical spectroscopic characterization and non-linear optical properties studies of a new non-centrosymmetric hybrid material. J. Mol. Struct. 1123 (2016) 144–152. DOI:10.1016/j.molstruc.2016.06.031 |
| [4] | N. Chihaoui, B. Hamdi, A. Ben Salah, R. Zouari, A new mononuclear complex:structure, vibrational (FT-IR and Raman), Hirshfeld surfaces analysis, electrical properties and equivalent circuit. J. Phys. Chem. Biophys 6 (2016) 216. |
| [5] | J. Berg, Potential metal-binding domains in nucleic acid binding proteins. Science 232 (1986) 485–487. DOI:10.1126/science.2421409 |
| [6] | R.H. Prince, P.R. Wolley, Metal ion function in carbonic anhydrase. Angew. Chem. Int. Ed. Engl. 11 (1972) 408–417. DOI:10.1002/(ISSN)1521-3773 |
| [7] | B. Luo, B.E. Kucera, W.L. Gladfelter, Syntheses and X-ray crystal structures of zinc complexes with an amido-diamine ligand. Polyhedron 25 (2006) 279–285. DOI:10.1016/j.poly.2005.06.041 |
| [8] | A. Scozzafava, L. Menabuoni, F. Mincione, G. Mincione, C.T. Supuran, Carbonic anhydrase inhibitors:synthesis of sulfonamides incorporating dtpa tails and of their zinc complexes with powerful topical antiglaucoma properties. Bioorg. Med. Chem. Lett. 11 (2011) 575–582. |
| [9] | G.V. Long, M.M. Harding, P. Turner, X-ray structure of the zinc complex of the central metal chelation site of the antitumour drug streptonigrin. Polyhedron 19 (2000) 1067–1071. DOI:10.1016/S0277-5387(00)00352-1 |
| [10] | L. Puccetti, G. Fasolis, D. Vullo, Ⅱ Ⅸ, and Ⅻ with Schiff's bases incorporating chromone and aromatic sulfonamide moieties, and their zinc complexes. Bioorg. Med. Chem. Lett. 15 (2005) 3096–3101. DOI:10.1016/j.bmcl.2005.04.055 |
| [11] | J. Kaizer, J. Pap, G. Speier, Synthesis, structure and catecholase activity of dinuclear copper and zinc complexes with an N3-ligand. J. Inorg. Biochem. 91 (2002) 190–198. DOI:10.1016/S0162-0134(02)00459-2 |
| [12] | O. Sénèque, M. Giorgi, O. Reinaud, Hydrogen bonding and CH/π interactions for the stabilization of biomimetic zinc complexes:first examples of X-ray characterized alcohol and amide adducts to a tetrahedral dicationic Zn center. Chem. Commun. (2001) 984–985. |
| [13] | S. Emami, S.J. Hosseinimehr, S.M. Taghdisi, S. Akhlaghpoor, Kojic acid and its manganese and zinc complexes as potential radioprotective agents. Bioorg. Med. Chem. Lett. 17 (2007) 45–48. DOI:10.1016/j.bmcl.2006.09.097 |
| [14] | J.H. Li, J.T. Wang, L.Y. Zhang, Structure, speciation, DNA binding and nuclease activity of two bipyridyl-zinc complexes bearing trimethylaminomethyl groups. Inorg. Chim. Acta 362 (2009) 1918–1924. DOI:10.1016/j.ica.2008.09.011 |
| [15] | A. Tarushi, G. Psomas, C.P. Raptopoulou, D.P. Kessissoglou, Zinc complexes of the antibacterial drug oxolinic acid:structure and DNA binding properties. J. Inorg. Biochem. 103 (2009) 898–905. DOI:10.1016/j.jinorgbio.2009.03.007 |
| [16] | J. Fielden, P.T. Gunning, D.L. Long, Anion control of isomerism, crystal packing and binding properties in a mononuclear zinc complex. Polyhedron 25 (2006) 3474–3480. DOI:10.1016/j.poly.2006.06.044 |
| [17] | W.S. Xia, C.H. Huang, D.J. Zhou, Photoelectric conversion from a hemicyanine dye containing zinc complex in a Langmuir-Blodgett film. Langmuir 13 (1997) 80–84. DOI:10.1021/la960108l |
| [18] | H. Firouzabadi, M. Adibi, B. Zeynizadeh, Modified borohydride agents; efficient reduction of azides with (1. 4-diazabicyclo[2.2.2] octane) (tetrahydroborato) zinc complex[Zn (BH4)2(Dabaco)] and methyltriphenylphosphonium tetrahydroborate[MePh3P+BH4-]. Synth. Commun. 28 (1998) 1257–1273. DOI:10.1080/00397919808005968 |
| [19] | R.Q. Fan, D.S. Zhu, Y. Mu, Syntheses, structures, and luminescent properties of[bis (iminoalkyl) pyridine]cadmium (Ⅱ) complexes. Eur. J. Inorg. Chem. 2004 (2004) 4891–4897. DOI:10.1002/(ISSN)1099-0682 |
| [20] | M. Amine Fersi, I. Chaabane, M. Gargouri, A. Bulou, Structure and characterization of the phase transition of the new organic-inorganic hybrid compound[C8H10NO]2[ZnCl4]. Polyhedron 85 (2015) 41–47. DOI:10.1016/j.poly.2014.08.056 |
| [21] | Y.M. Xie, W.T. Chen, J.H. Wu, Synthesis structure, and physical properties of[Sm (C6NO2H5)3(H2O)2]2n.(H5O2)n(ZnCl5)n(ZnCl4)2n·(H2O)2n with unprecedented ZnCl53- species. J. Solid State Chem. 181 (2008) 1853–1858. DOI:10.1016/j.jssc.2008.04.002 |
| [22] | S.R. Ali Khan, S.R. Huang, S. Shamsuddin, Synthesis, characterization and cytotoxicity of new platinum (Ⅳ) axial carboxylate complexes:crystal structure of potential antitumor agent[PtⅣ(trans-1R, 2R-diaminocyclohexane) trans (acetate)2Cl2]. Bioorg. Med. Chem. 8 (2000) 515–521. DOI:10.1016/S0968-0896(99)00313-2 |
| [23] | S. Nayab, H. Lee, J.H. Jeong, Synthesis and structural characterization of a dichloro zinc complex of N, N'-bis-(2, 6-dichloro-benzyl)-(R, R)-1, 2-diaminocyclohexane:application to ring opening polymerization of raclactide. Polyhedron 31 (2012) 682–687. DOI:10.1016/j.poly.2011.10.035 |
| [24] | Y.Y. Sun, G. Xu, Z. Cao, S.H. Gou, Synthesis and biological evaluation of platinum (Ⅱ) complexes containing (1R, 2R)-N1-alkyl-1, 2-diaminocyclohexane and D-(+)-camphorate ligands. Inorg. Chim. Acta 395 (2013) 154–159. DOI:10.1016/j.ica.2012.10.011 |
| [25] | J. Fang, X. Wei, J.B. Sapp, Y.J. Deng, Novel platinum (Ⅱ) complexes containing diaminocyclohexane and thiourea derivative ligands:synthesisand X-ray crystal structure of (trans-1, 2-diaminocyclohexane) dithioureaplatinum (Ⅱ) nitrate monohydrate. Inorg. Chim. Acta 411 (2014) 5–10. DOI:10.1016/j.ica.2013.11.012 |
| [26] | I. Mkaouar, B. Hamdi, N. Karâa, R. Zouari, Synthesis, solid-state characterization and dielectric properties of a trichlorostanate (Ⅱ) complex. Polyhedron 87 (2015) 424–432. DOI:10.1016/j.poly.2014.10.035 |
| [27] | A. Valkonen, K. Ahonen, E. Kolehmainen, Bis (6-thioxo-1, 6-dihydropurinium) tetrachlorozincate (Ⅱ). Acta Cryst. C 62 (2006) m290–m292. DOI:10.1107/S0108270106019469 |
| [28] | B.M. Craven, G.L. Gartland, The 2:1 crystal complex of 55-diethylbarbituric acid (barbital) and caffeine. Acta Cryst. 30 (1974) 1191–1195. DOI:10.1107/S0567740874004559 |
| [29] | M.A. Spackman, J.J. McKinnon, Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 4 (2002) 378–392. DOI:10.1039/B203191B |
| [30] | M.A. Spackman, P.G. Byrom, A novel definition of a molecule in a crystal. Chem. Phys. Lett. 267 (1997) 215–220. DOI:10.1016/S0009-2614(97)00100-0 |
| [31] | J.J. McKinnon, M.A. Spackman, A.S. Mitchell, Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Cryst. 60 (2004) 627–668. DOI:10.1107/S0108768104020300 |
| [32] | J.J. McKinnon, D. Jayatilaka, M.A. Spackman, Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. (2007) 3814–3816. |
| [33] | J. Sivaraman, K. Subramanian, D. Velmurugan, E. Subramanian, K. Balakrishna, Structure of vicogenin. Acta Cryst. C49 (1993) 1240–1242. |
| [34] | M.A. Spackman, D. Jayatilaka, Hirshfeld surface analysis. CrystEngComm 11 (2009) 19–32. DOI:10.1039/B818330A |
| [35] | D. Massiot, F. Fayon, M. Capron, Modelling one-and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 40 (2002) 70–76. DOI:10.1002/mrc.984 |
| [36] | L.E. Ericson, J.E. Sarneski, C.N. Reilley, Carbon-13 nuclear magnetic resonance studies of platinum (Ⅱ) complexes. I. Five-membered rings formed by substituted 1, 2-diaminoethanes. Inorg. Chem. 14 (1975) 307–3017. |
| [37] | N. Karâa, B. Hamdi, A. Ben Salah, R. Zouari, Synthesis Infra-red, CP/MAS-NMR characterization, structural study and electrical properties of thebis (4-amino-2-chloropyridinium) tetrachlorozincate (Ⅱ) monohydrate. J. Mol. Struct. 1049 (2013) 48–58. DOI:10.1016/j.molstruc.2013.06.003 |
| [38] | A. Samet, H. Boughzala, H. Khemakhem, Y. Abid, Synthesis, characterization and non-linear optical properties of Tetrakis (dimethylammonium) Bromide Hexabromobismuthate:{[(CH3)2NH2]+}4·Br-·[BiBr6]3-. J. Mol. Struct. 984 (2010) 23–29. DOI:10.1016/j.molstruc.2010.08.049 |
| [39] | G. Mahalakshmi, V. Balachandran, Molecular structure, vibrational spectra (FTIR and FT Raman) and natural bond orbital analysis of 4-Aminomethylpiperidine:DFT study. Spectrochim. Acta A Mol. Biomol. Spectrosc. 131 (2014) 587–598. DOI:10.1016/j.saa.2014.04.154 |
| [40] | D. Sajan, J. Binoy, B. Pradeep, NIR-FT Raman and infrared spectra and ab initio computations of glycinium oxalate. Spectrochim. Acta A 60 (2004) 173–180. DOI:10.1016/S1386-1425(03)00193-8 |
| [41] | Y.B. Shankar Rao, M.V.S. Prasad, N. Udaya Sri, V. Veeraiah, Vibrational (FT-IR, FT-Raman) and UV-visible spectroscopic studies, HOMO-LUMO, NBO, NLO and MEP analysis of Benzyl (imino (1H-pyrazol-1-yl) methyl) carbamate using DFT calculations. J. Mol. Struct. 1108 (2016) 567–582. DOI:10.1016/j.molstruc.2015.12.008 |
| [42] | H.J. Schütt, A new phenomenological description of the electrical relaxation in ionic conductors. Solid State Ionics 72 (1994) 86–88. DOI:10.1016/0167-2738(94)90129-5 |
| [43] | I.M. Hodge, C.A. Angell, Electrical relaxation in amorphous protonic conductors. J. Chem. Phys. 67 (1977) 1647. DOI:10.1063/1.434997 |
| [44] | F. Alvarez, A. Alegíia, J. Colmenero, Interconnection between frequencydomain Havriliak-Negami and time-domain Kohlrausch-Williams-Watts relaxation functions. Phys. Rev. B 47 (1993) 125–130. DOI:10.1103/PhysRevB.47.125 |
| [45] | Z. Yu, C. Ang, Maxwell-Wagner polarization in ceramic composites BaTiO3-(Ni0.3Zn0.7) Fe2.1O4. J. Appl. Phys. 91 (2002) 794. DOI:10.1063/1.1421033 |
| [46] | S.S. Shinde, C.H. Bhosale, K.Y. Rajpure, Studies on morphological and electrical properties of Al incorporated combusted iron oxide. J. Alloys Compounds 509 (2011) 3943–3951. DOI:10.1016/j.jallcom.2010.12.185 |
| [47] | G. Hutchins, O. Abu-Alkhair, M.M. El-Nahass, K. Abdel-Hady, Electrical conductivity and dielectric relaxation in non-crystalline films of tungstentrioxide. J. Non-Cryst. Solids 353 (2007) 4137–4142. DOI:10.1016/j.jnoncrysol.2007.06.042 |
| [48] | K. Prasad, Li ly, K. Kumari, K.L. Yadav, Hopping type of conduction in (Na0.5Bi0.5) ZrO3 ceramic. J. Phys. Chem. Solids 68 (2007) 1508–1514. DOI:10.1016/j.jpcs.2007.03.023 |
| [49] | J. Tarasiewicz, R. Jakubas, J. Zaleski, J. Baran, Structural characterization, thermal, dielectric and spectroscopic properties of di (n-pentylammonium) pentabromoantimonate (Ⅲ):. J. Mol. Struct. 876 (2008) 86–101. DOI:10.1016/j.molstruc.2007.06.005 |
| [50] | A. Bhan, Y.V. Joshi, W.N. Delgass, K.T. Thomson, DFT investigation of alkoxide formation from olefins in H-ZSM-5. J. Phys. Chem. B 107 (2003) 10476–10487. DOI:10.1021/jp034382h |
| [51] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., PopleGaussian 03, Revision C.02, Gaussian Inc., Wallingford CT, 2004. |
| [52] | P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82 (1985) 299. DOI:10.1063/1.448975 |
| [53] | A.D. Becke, Density-functional thermochemistry. Ⅲ. The role of exact exchange. J. Chem. Phys. 98 (1993) 5648. DOI:10.1063/1.464913 |
| [54] | R. Dennington, T. Keith, J. Millam, Gauss View Version 5, Semichem Inc., Shawnee Mission, KS, 2009. |
| [55] | L.J. Farrugia, WinGXsuite for small-molecule single-crystal crystallography. J. Appl. Cryst. 32 (1999) 837–838. DOI:10.1107/S0021889899006020 |
| [56] | G.M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1997. |
| [57] | G.M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997. |
| [58] | L.J. Farrugia, ORTEP-3 for windows-aversionof ORTEP-Ⅲ with a GraphicalUser Interface (GUI). J. Appl. Cryst. 30 (1997) 565. |
| [59] | K. Brandenburg, Diamond Version 2.0, ImpactGbR, Bonn, Germany, 1998. |