 2017, Vol. 28
2017, Vol. 28 



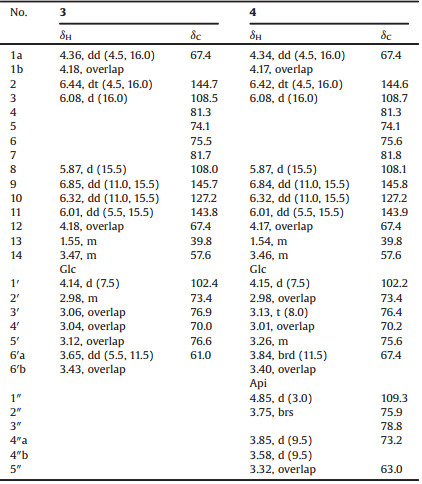
Medicinal plants supply a rich source for screening bioactive constituents. As the common traditional Chinese medicine, Atractylodes lancea has been reputed for "strengthening spleen, removing cold, and improving eyesight" [1, 2]. Previous researches reported a series of steroids, monoterpenes, polyacetylenes, and sesquiterpenoids, which were mainly isolated from the petroleum ether and ethyl acetate extract of this plant [3-8]. In our continuing phytochemical investigations on the n-BuOH part of an aq. EtOH extract of A. lancea [9], one new eudesmane-type sesquiterpenoid glycoside (1), one new guaiane-type sesquiterpenoid glycoside (2), and two new C14-polyacetylene glycosides (3, 4) (Fig. 1) were isolated using various column chromatographic methods. Their structures were identified on the basis of spectrometric and spectroscopic data. The configurational assignments of 1-4 were established using experimental and calculated electronic circular dichroism (ECD), whereas those of monosaccharide moieties were determined by GC after the chiral derivatization. All the compounds were assayed for anti-inflammatory effects on the lipopolysaccharide (LPS)-induced NO production in microglia BV2 cells.

|
Download:
|
| Figure 1. The chemical structures of compounds 1–4 | |
2. Results and discussion
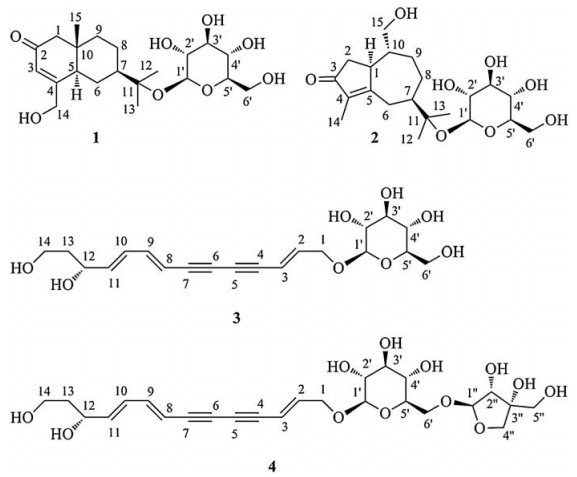
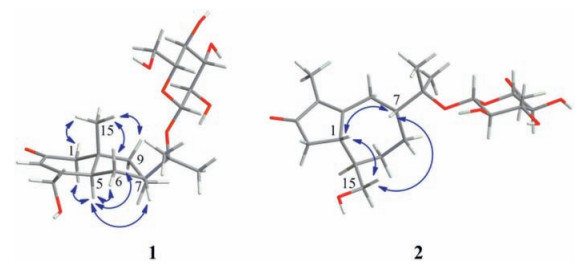
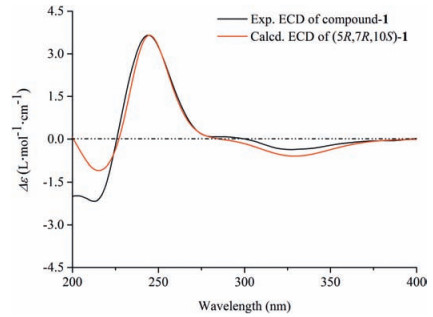
Compound 1 was assigned the molecular formula C21H34O8 on the basis of the HR-ESIMS adduct ion at m/z 437.2155 [M+Na]+, suggesting five degrees of unsaturation. Its IR absorption at υmax 1654 cm-1 showed the presence of an α, β-unsaturated ketone unit, which was confirmed by the resonances at δC 121.2, 167.0, and 198.2 in the 13C NMR spectrum (Table 1). Except for the β-D-glucopyranosyl moiety (97.1, 73.7, 76.6, 70.3, 77.0, and 61.4), the remaining 15 carbons revealed a skeleton of eudesmane-type sesquiterpenoid, whose 1D NMR data (Table 1) were highly similar to those of 14-hydroxyisopterocarpolone [10]. The key HMBC cross-peak (Fig. 2) of H-1' with C-11 revealed the location of the glucosyl moiety at C-11. Therefore, the structure of 1 was elucidated as 14-hydroxyisopterocarpolone-11-O-β-D-glucopyranoside, as evidenced by further analyses of other long-range correlations. In the ROESY experiment (Fig. 3), correlations of H-5 with H-1a, H-6a, H-7, and H-9b and those of H3-15 with H-1b, H-6b, and H-9a indicated that the juncture of A-and B-rings was trans-configured, H-5 and CH3-10 was axial, whereas C-7 hydroxyisopropyl was equatorial. Based on the Snatzke's rule for the cyclohexenone unit [11, 12], the negative (Δε=-0.37) Cotton effect (CE) at 327 nm contributed by the n→π* electron transition of the α, β-unsaturated ketone unit unambiguously favored a (5R, 7R, 10S) configuration. This configurational assignment was further supported by the ECD calculation, which was perfomed using an MMFF94 force field and the time-dependent density functional theory (TDDFT) at the B3LYP/6-31 +G (d, p) level (see Supplementary data, S22). As a result, the calculated spectrum (Fig. 4) of (5R, 7R, 10S)-diastereomer well matched with the experimental spectrum for compound 1.
|
|
Table 1 1H NMR (500 MHz) and 13C NMR (125 MHz) data for compounds 1 and 2 in DMSOd6. |


|
Download:
|
| Figure 2. Key HMBC and 1H–1H COSY correlations of compounds 1, 2, and 4 | |

|
Download:
|
| Figure 3. Key ROESY correlations of compounds 1 and 2 | |

|
Download:
|
| Figure 4. Experimental and calculated ECD spectra of compound 1 | |
The HR-ESIMS of compound 2 showed an adduct ion peak at m/z 415.2339 [M+ H]+, whose molecular formular was deduced as C21H34O8 (calcd. for 415.2332). IR apsorption at 1680 cm-1 was indicative of a conjugated carbonyl group. The 13C NMR data (Table 1) confirmed the presence of an α, β-unsaturated ketone unit (δC 134.3, 177.4, and 207.8), a β-D-glucopyranosyl moiety (δC 97.0, 73.9, 77.2, 70.4, 76.6, and 61.3), three primary carbons (δC 7.6, 21.6, and 24.4), five second carbons (δC 26.2, 26.6, 28.3, 41.3, and 65.4), as well as four tertiary carbons (βC 44.8, 45.8, 46.3, and 79.1). Based on the aforementioned evidences, the distinctive four methyl groups (βH 1.15, 1.16, and 1.60) and a series of aliphatic protons allowed us to propose a skeleton of guaiane-type sesquiterpenoid with a β-D-glucopyranosyl moiety at C-11 [13]. Detailed analyses of the HSQC and HMBC data (Fig. 2) established the structure of 2 as 11, 15-dihydroxy-4-guaien-3-one 11-O-β-Dglucopyranoside. The ROESY correlations (Fig. 3) of H-1 with H-7/H2-15 and that of H-7 with H2-15 suggested that H-1, H-7, and H2-15 were on the same side of the guaiane-ring. To designate the stereochemistry, the ECD caculation was perfomed using an MMFF94 force field and the TDDFT method at the B3LYP/6-31 + G (d, p) level (see Supplementary data, S22). The theoretical data (Fig. 5) of (1S, 7R, 10R)-diastereomer was well in agreement with the experimental data for 2.

|
Download:
|
| Figure 5. Experimental and calculated ECD spectra of compound 2 | |
Compounds 3 and 4 were deduced to have a similar skeleton of an ene-diyne-diene chromophore by HPLC-DAD analyses [14, 15]. Their molecular formulas were established as C20H26O8 and C25H34O12 by the HR-ESIMS adduct ions at m/z 439.1625 [M +COOH]- and 525.1980 [M-H]-, respectively. For compound 3, in addition to a β-D-glucopyranosyl moiety (δC 102.4, 73.4, 76.9, 70.0, 76.6, and 61.0), the 13C NMR data (Table 2) revealed the presence of two acetylenic bonds at δC 74.1, 75.5, 81.3, and 81.7, three olefinic bonds at δC 108.0, 108.5, 127.2, 143.8, 144.7, and 145.7, two primary carbons at δC 57.6 and 67.4, and two secondary carbons at δC 39.8 and 67.4. The three olefinic bonds were further attributed to be trans-configured by the 3JH-H values (16.0 Hz, 15.5 Hz, and 15.5 Hz). The above 1D NMR spectral features coupled with the information obtained from the 1H-1H COSY and HMBC spectra suggested the attachments of a C (12) H-C (13) H2-C (14) H2 chain at C-11 and a glucosyl moiety at C-1. Therefore, the structure of 3 was elucidated as (2E, 8E, 10E)-tetradeca-2, 8, 10-triene-4, 6-diyne-1, 12, 14-triol-1-O-β-D-glucopyranoside. Compound 4 was determined to have an additional apiofuranosyl moeity (δC 109.3, 75.9, 78.8, 73.2, and 63.0) on the basis of the 13C NMR data (Table 2). A deshielded carbon resonance at δC 67.4 indicated that the apiosyl moiety was substituted at Glc-C-6'. This assignment was verified by HMBC analyses (Fig. 2). Thus, the structure of compound 4 was establsihed as shown in Fig. 1. The absolute configurations of 3 and 4 were determined by the ECD caculations using an MMFF94 force field and TDDFT method at the B3LYP/6-31 + G (d, p) level (see Supplementary data, S22). The calculated spectrum of (12R)-diastereomer was well in agreement with the experimental spectra of 3 and 4. Consequently, their absolute configurations were designated as 12R (Fig. 6).
|
|
Table 2 1H NMR (500 MHz) and 13C NMR (125 MHz) data for compounds 3 and 4 in DMSOd6. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Figure 6. Experimental and calculated ECD spectra of compounds 3 and 4 | |
{kind=link}
All compounds were tested for the anti-inflammatory effects on the LPS-induced NO production in the microglia BV2 cells at a concentration of 10 μmol L-1. Compound 4 showed weak antiinflammatory effect with the inhibitory ratios of 19.60% (curcumin with 72.50%).
3. ConclusionPhytochemical investigation of the rhizomes of A. lancea led to the isolation of two new sesquiterpenoid glycosides (1, 2) and two new C14-polyacetylene glycosides (3, 4) (Fig. 1). The configurational assignments of their aglycones were established by comparing the experimental and calculated ECD, whereas those of monosaccharide moieties were determined by GC after the chiral derivatization. Compound 4 exhibited weak anti-inflammatory effect. The obtained results were benefit for the subsequent phytochemical researches of genus Atractylodes.
4. Experimental 4.1. General proceduresOptical rotations were measured on a JASCO P-2000 automatic polarimeter. UV spectra were obtained on a JASCO V-650 spectrophotometer. ECD spectra were recorded on a JASCO J-815 spectropolarimeter. IR spectra were achieved on a Nicolet 5700 spectrometer using an FT-IR microscope transmission method. NMR experiments were conducted on a Bruker spectrometer (500 MHz). Chemical shifts were given with the solvent peaks as the references (δ in ppm, J in Hz). HR-ESIMS data were obtained using an Agilent 1100 series LC/MSD TOF from Agilent Technologies. GC experiments were conducted on an Agilent 7890A instrument. Column chromatography was performed with macroporous resin (Diaion HP-20, Mitsubishi Chemical Corp., Tokyo, Japan), RP-18 (50 μm, YMC, Kyoto, Japan) and Sephadex LH-20 (Pharmacia Fine Chemicals, Uppsala, Sweden). A Shimadzu LC-10AT instrument equipped with an YMC-Pack ODS-A column (250 × 20 mm, 5 mm, Japan) served as the reversed-phase preparative HPLC. HPLC analyses were performed on an Agilent 1260 series system with an Apollo C18 column (250 × 4.6 mm, 5 mm, Grace Davison).
4.2. Plant materialsThe rhizomes of A. lancea were collected in Huanggang City, Hubei Province, China, in June 2014 and authenticated by Prof. L. Ma. A voucher specimen (ID-s-2596) was deposited in the herbarium at the Department of Medicinal Plants, Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing 100050, China.
4.3. Extraction and isolationThe dried rhizomes of A. lancea (100 kg) were extracted thrice with 80% EtOH (v/v) under reflux condition for 2 h. The crude extract (25.6 kg) was suspended in 30 L distilled H2O and separately partitioned with petroleum ether, EtOAc, and n-BuOH (three times each). The n-BuOH fraction (1.2 kg) was chromatographed on an HP-20 column and eluted with a step gradient of EtOH-H2O (v/v) to provide five fractions: A (H2O fraction, 824 g), B (15% EtOH fraction, 88.6 g), C (30% EtOH fraction, 106.4 g), D (50% EtOH fraction, 53.3 g), and E (95% EtOH fraction, 19.5 g). Fraction C was chromatographed on an RP-18 column and eluted at a gradient of MeOH-H2O (0:100-100:0, v/v) to obtain fractions C1-C10 using HPLC analyses. Fraction C4 (10.2 g) was chromatographed on an LH-20 column using H2O to obtain 32 subfractions (Fr. C4.1-Fr. C4.32). Subtractions Fr. C4.5-Fr. C4.6 were further separated using the reversed-phase preparative HPLC, with 30% MeOH (v/v) to yield 2 (4.9 mg). Fraction C5 (12.9 g) was eluted on an LH-20 column using H2O to yield 38 subfractions (Fr. C5.1-Fr. C5.38). Fr. C5.35-Fr. C5.38 were purified using the reversed-phase preparative HPLC with a MeOH:H2O ratio of 35:65 (v/v) to afforded 3 (7.6 mg). Fraction C6 (6.3 g) was chromatographed on an LH-20 column with H2O to produce 37 subfractions (Fr. C6.1-Fr. C6.37). Fr. C6.11-Fr. C6.14 were purified using the reversed-phase preparative HPLC with 35% MeOH (v/v) to produce 1 (5.8 mg), similarly, Fr. C6.35-Fr. C6.36 yielded 4 (14.8 mg).
4.4. (5R, 7R, 10S)-14-Hydroxyisopterocarpolone-11-O-b-Dglucopyranoside (1)White amorphous powder; [α]D20 + 52.7 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 240 (3.87) nm; ECD (MeOH) λmax (Δε) 213 (-2.17), 244 (+3.65), 327 (-0.37) nm; IR (KBr) υmax 3359, 2937, 1654, 1078, 1031 cm-1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 437.2155 [M+Na]+ (calcd for C21H34O8Na, 437.2151).
4.5. (1S, 7R, 10R)-11, 15-Dihydroxy-4-guaien-3-one 11-O-b-Dglucopyranoside (2)White amorphous powder; [α]D20 -32.5 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 248 (4.11) nm; ECD (MeOH) λmax (Δε) 235 (-0.26), 264 (-0.62), 298 (-0.78) nm; IR (KBr) υmax 3360, 2973, 2923, 1680, 1079, 1044 cm-1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 415.2339 [M+H]+ (calcd. for C21H35O8, 415.2332).
4.6. (2E, 8E, 10E, 12R)-Tetradeca-2, 8, 10-triene-4, 6-diyne-1, 12, 14-triol-1-O-b-D-glucopyranoside (3)Brown amorphous powder; [α]D20 -149.6 (c 0.05, MeOH: H2O=1:1); UV (MeOH) λmax (log ε) 207 (4.22), 250 (4.08), 296 (4.06), 315 (4.15), 337 (4.03) nm; ECD (MeOH: H2O=1:1) λmax (Δε) 216 (-0.42) nm; IR (KBr) υmax 3367, 2925, 2886, 2198, 2128, 1634, 1078, 1045 cm-1; 1H and 13C NMR data, see Table 2; HR-ESIMS m/z 439.1625 [M+COOH]- (calcd. for C21H27O10, 439.1604).
4.7. (2E, 8E, 10E, 12R)-Tetradeca-2, 8, 10-triene-4, 6-diyne-1, 12, 14-triol-1-O-b-D-apiofuranosyl-(1! 6)-b-D-glucopyranoside (4)Brown amorphous powder; [α]D20 -27.6 (c 0.05, MeOH: H2O=1:1); UV (MeOH) λmax (log ε) 212 (3.94), 252 (3.94), 296 (3.89), 315 (3.98), 337 (3.83) nm; ECD (MeOH: H2O=1:1) λmax (Δε) 211 (-1.36), 241 (-0.24) nm; IR (KBr) υmax 3374, 2885, 2198, 1636, 1057 cm-1; 1H and 13C NMR data, see Table 2; HR-ESIMS m/z 525.1980 [M-H]- (calcd. for C25H33O12, 525.1972).
4.8. Sugar analysesCompounds 4 (3 mg) was hydrolyzed with 1 mol L-1 HCldioxane (v/v=1:1, 5 mL) at 60 ℃ for 6 h [16]. The obtained hydrolysates were extracted with EtOAc three times (3 mL) to yield EtOAc extracts and monosaccharide residues after evaporating the solvents. The monosaccharide residues were processed based on the reported method [17-19]. The absolute configurations of glucose and apiose were determined by comparing the retention times of their trimethylsilyl-L-cysteine derivatives with those of authentic sugars prepared by a similar procedure: D-glucose at 20.56 min, D-apiose at 14.56 min (see Supplementary data, S21).
4.9. Anti-inflammatory activity assayThe anti-inflammatory activity assay was tested in the microglial BV2 cell (curcumin as the positive drug). The detailed processes were based on the reported method [9], which were also presented in the "supplementary data" (S23).
AcknowledgmentThe authors are grateful to Y. H. Wang, M. X. Jin, and X. Liu for conducting the NMR experiments. The work described in this publication was funded by the National Science and Technology Project of China (No. 2012ZX09301002-001003).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.10.036.
| [1] | ${referAuthorVo.mingEn} Chinese Pharmacopoeia Commission, Chinese Pharmacopoeia, vol.Ⅰ, Beijing: China Medical Science Press, 2015 : 161 -162. |
| [2] | N. Koonrungsesomboon, K. Na-Bangchang, J. Karbwang, Therapeutic potential and pharmacological activities of Atractylodes lancea (Thunb.) DC. Asia Pac. J. Trop. Med. 7 (2014) 421–428. DOI:10.1016/S1995-7645(14)60069-9 |
| [3] | H. Meng, G.Y. Li, R.H. Dai, Chemical constituents of Atractylodes chinensis (DC.) Koidz. Biochem. Syst. Ecol. 38 (2010) 1220–1223. DOI:10.1016/j.bse.2010.12.023 |
| [4] | M. Resch, A. Steigel, Z.L. Chen, R. Bauer, 5-Lipoxygenase and cyclooxygenase-1 inhibitory active compounds from Atractylodes lancea. J. Nat. Prod. 61 (1998) 347–350. DOI:10.1021/np970430b |
| [5] | M. Resch, J. Heilmann, A. Steigel, R. Bauer, Further phenols and polyacetylenes from the rhizomes of Atractylodes lancea and their anti-inflammatory activity. Planta Med. 67 (2001) 437–442. DOI:10.1055/s-2001-15817 |
| [6] | H.X. Wang, C.M. Liu, Q. Liu, K. Gao, Three types of sesquiterpenes from rhizomes of Atractylodes lancea. Phytochemistry 69 (2008) 2088–2094. DOI:10.1016/j.phytochem.2008.04.008 |
| [7] | M.S. Lehner, A. Steigel, R. Bauer, Diacetoxy-substituted polyacetylenes from Atractylodes lancea. Phytochemistry 46 (1997) 1023–1028. DOI:10.1016/S0031-9422(97)00342-7 |
| [8] | H. Meng, G.Y. Li, R.H. Dai, Two new polyacetylenic compounds from Atractylodes chinensis (DC.) Koidz. J. Asian Nat. Prod. Res. 13 (2011) 346–349. DOI:10.1080/10286020.2011.557662 |
| [9] | K. Xu, J.S. Jiang, Z.M. Feng, Bioactive sesquiterpenoid and polyacetylene glycosides from Atractylodes lancea. J. Nat. Prod. 79 (2016) 1567–1575. DOI:10.1021/acs.jnatprod.6b00066 |
| [10] | H. Kamauchi, K. Kinoshita, K. Takatori, New sesquiterpenoids isolated from Atractylodes lancea fermented by marine fungus. Tetrahedron 71 (2015) 1909–1914. DOI:10.1016/j.tet.2015.02.041 |
| [11] | G. Snatzke, Circulardichroismus-Ⅸ:modifizierung der octantenregel für α, β-ungesättigte ketone:transoide enone. Tetrahedron 21 (1965) 421–438. DOI:10.1016/S0040-4020(01)98283-3 |
| [12] | J.K. Gawronski, Circular dichroism and stereochemistry of chiral conjugated cyclohexenones. Tetrahedron 38 (1982) 3–26. DOI:10.1016/0040-4020(82)85040-0 |
| [13] | Y. Yu, H. Gao, Y. Dai, Guaiane-type sesquiterpenoid glucosides from Gardenia jasminoides Ellis. Magn. Reson. Chem. 49 (2011) 258–261. DOI:10.1002/mrc.v49.5 |
| [14] | J. He, Y. Shen, J.S. Jiang, New polyacetylene glucosides from the florets of Carthamus tinctorius and their weak anti-inflammatory activities. Carbohydr. Res. 346 (2011) 1903–1908. DOI:10.1016/j.carres.2011.06.015 |
| [15] | J.Y. He, S. Zhu, K. Komatsu, HPLC/UV analysis of polyacetylenes, phenylpropanoid and pyrrolidine alkaloids in medicinally used Codonopsis species. Phytochem. Anal. 25 (2014) 213–219. DOI:10.1002/pca.v25.3 |
| [16] | M.L. Gan, M.T. Liu, L.S. Gan, Dammarane glycosides from the root of Machilus yaoshansis. J. Nat. Prod. 75 (2012) 1373–1382. DOI:10.1021/np300310a |
| [17] | S. Hara, H. Okabe, K. Mihashi, Gas-liquid chromatographic separation of aldose enantiomers as trimethylsilyl ethers of methyl 2-(polyhydroxyalkyl)-thiazolidine-4-(R)-carboxylates. Chem. Pharm. Bull. 35 (1987) 501–506. DOI:10.1248/cpb.35.501 |
| [18] | Z.M. Feng, S. Song, J. He, Acyl glycosides with rare β-D-apiofuranosyl-β-D-glucopyranosyl-β-D-apiofuranosyl from Erycibe hainanesis. Carbohydr. Res. 380 (2013) 59–63. DOI:10.1016/j.carres.2013.07.011 |
| [19] | Y. Shen, Z.M. Feng, J.S. Jiang, Y.N. Yang, P.C. Zhang, Dibenzoyl and isoflavonoid glycosides from Sophora flavescens:inhibition of the cytotoxic effect of Dgalactosamine on human hepatocyte HL-7702. J. Nat. Prod. 76 (2013) 2337–2345. DOI:10.1021/np400784v |