 2017, Vol. 28
2017, Vol. 28 


 , Xiao-Mei Langa, Lian-Ming Yangb, Sheng-Yuan Zhoua, Hong-Fan Hua, Shan Xuea, Xin Suna, Shi-Xuan Xina
, Xiao-Mei Langa, Lian-Ming Yangb, Sheng-Yuan Zhoua, Hong-Fan Hua, Shan Xuea, Xin Suna, Shi-Xuan Xina
b Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Green Printing, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
N-Aryl substituted indenoindole compounds are attractive for chemical synthesis because of their importance in preparation of metallocene catalysts for the polymerization of ethylene and propylene [1, 2]. Metallocene catalyst has been modified worldwide in industrial and academic areas to provide many different catalyst-structures that can be used to synthesize highly isotactic, syndiotactic, atactic, hemi-isotactic polyolefin with different molecular weights and different degrees of tacticity [3-8]. N-Aryl substituted indenoindole derivatives are important building block for the ancillary ligands of metallocene catalyst owning to their cyclopentadiene functional group. Until now, rare reports have been found involving the synthesis of N-aryl substituted indenoindole [2], which also restrict the rational design of metallocene catalyst structures.
Nickel-catalyzed aromatic C--N coupling reactions have received intense investigations, and have become important and powerful tools in organic syntheses during the past decade [9-15]. The Ullmann reaction [16] is a classical method for the Cu-mediated C--N coupling and recently, the more practical ligand-promoted Cu-catalyzed chemistry [17-19] (so-called postUllmann reaction) has also been developed. Compared to the corresponding Pd and Cu-catalyzed systems, the major advantages of Ni-based catalysts are their much lower cost and potentially high reactivity toward easily-available but relatively inert substrates (e.g., aryl chlorides and aryl bromide) without the use of specially tailored organic ligands. Some works on the synthesis of N-aryl substituted indenoindole derivatives via cross coupling of aromatic halogen and indenoindole under metal catalysis has been done, the result disclosed the fact that little or none desired product was received [20]. Yang and co-workers demonstrated a number of nickel-catalyzed triarylamine synthesis via the amination of bromo-/iodoarene with metal diarylamides, in-situ produced from a diarylamine and the Grignard reagent or sodium hydride [21-25]. They proposed that simple anilines are not over arylated and the reaction proceeds "selectively" to the monosubstitution stage, even in the presence of excess aryl halides, indicating that a diarylamine is not coupled with an aryl halide under the "normal" conditions (i.e., typically a combination of free amines and inorganic bases). Accordingly, the N-aryl substituted indenoindole and the triarylamine have the similar conjugate structure, therefore the synthesis of N-aryl substituted indenoindole is a challenging task. The reason was presumably that the indenoindole is usually difficult to coordinate to the Ni center due to its steric bulk and poor nucleophilicity against a relatively small Ni center. For the metal-catalyzed coupling reaction, a coordination process, namely transmetalation, is one of the key steps in a catalytic cycle. Based on the situation, butyl lithium was selected as the base for a model reaction between bromobenzene and indenoindole. Indenoindole is readily deprotonated by butyl lithium and transformed to the more nucleophilic amide species, which enhances its ability to bind Ni center, and in addition, the use of highly air-sensitive nickel (0) precatalysts is avoided because easy-to-handle nickel (Ⅱ) compounds can be reduced to the lowvalent nickel species by butyl lithium agents in situ. Thus we proposed to employ the butyl lithium as the strong base to generate nucleophilic amides and as the reduction agent to produce low-valent Ni species in situ.
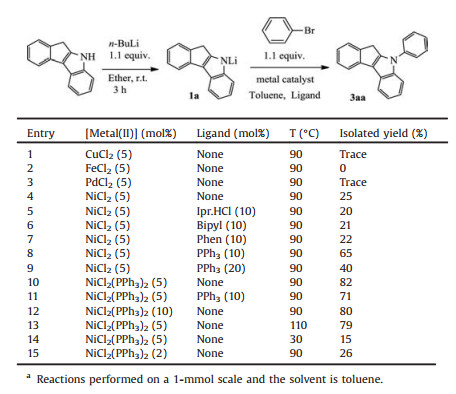
2. Results and discussionThe effect of the bases employed was evaluated first. Indeed, those commonly used bases in metal-catalyzed C--N coupling do not work. For instance, no expected coupling product was observed when sodium tert-butoxide, potassium tert-butoxide, or potassium orthophosphate were used. Sodium hydride, although it is able to convert indenoindole into the more nucleophilic amide species, is also ineffective in facilitating the coupling reaction. In an exploring model reaction, butyl lithium was used as the double function reagent, i.e. as a strong base to generate the amide and also as a reducing agent for the reduction of nickel salt to the low valent species. In the model indenoindole/bromobenzene coupling reaction, a brief set of screening experiments were focused on different metal catalyst systems and the results are summarized in Table 1. Several common transition metal (Ⅱ) precatalysts were surveyed, and it was found that only nickel showed the capability of promoting the reaction compared to others, such as iron, copper and palladium (entries 1-4). Toluene turned out to be the suitable solvent for the model reaction compared with other solvents tested. Nickel catalysts bearing phosphine ligands performed better than those of other ligand types (entry 8 vs. entries 5-7), and triphenylphosphine (PPh3) ligand offers relatively high activity (entry 8). To our pleasure, successful coupling in the model reaction resulted in 65% yield of the desired product when 2PPh3/nickel combination as the catalyst. Further increase the PPh3/Ni ratio to 4/1 was detrimental (entry 9). However, excellent coupling yield (82%, entry 10) was obtained when NiCl2(PPh3)2 complex was employed without any additional free triphenylphosphine. Noted also that the use of additional free PPh3 dose not help to enhance the catalytic efficiency (entry 11). Increasing the NiCl2(PPh3)2 catalyst loading does not improve the coupling yield, and evidenced dropping in coupling yield also occurred when the catalyst loading was decreased (entries 12 and 15). Higher temperature could not enhance the coupling reaction (entry 13) though, the reaction at room temperature gave only a low product yield of 15% (entry 14). Therefore the optimal conditions for this coupling reaction were set as in entry 10 of Table 1.
|
|
Table 1 Screening of catalyst systems.a |
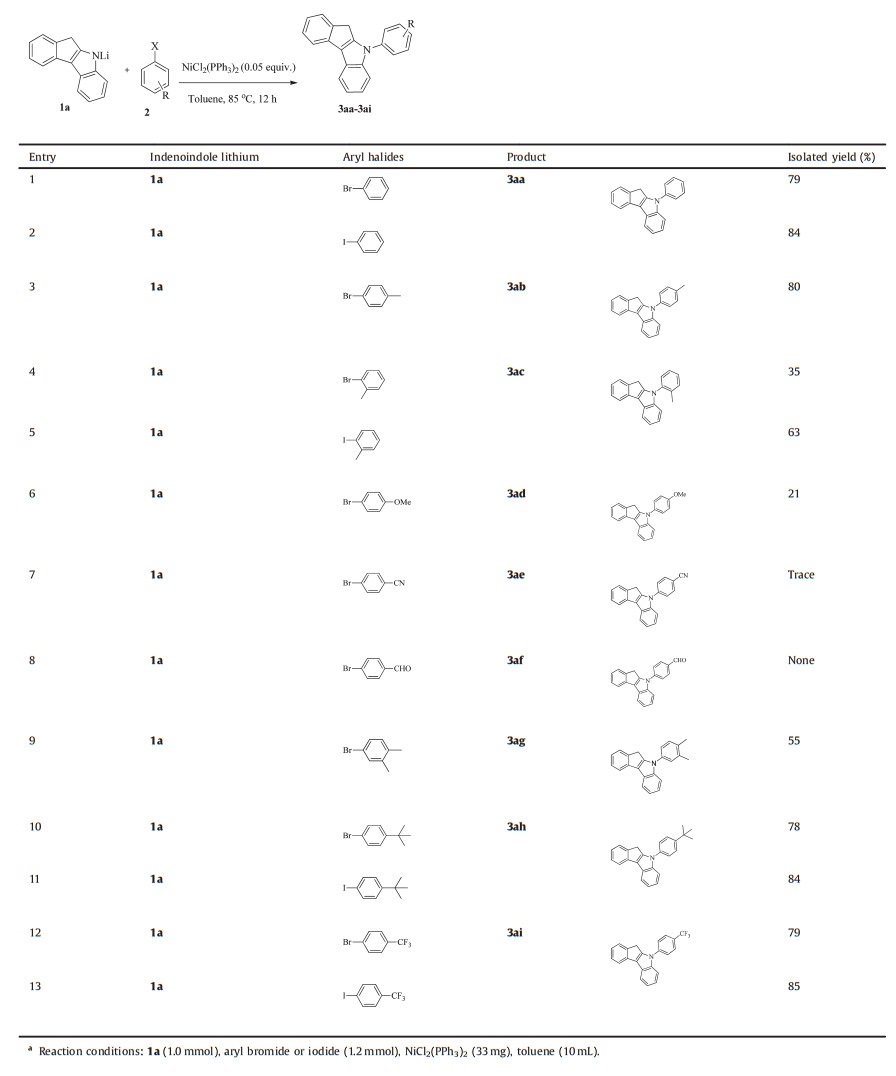
The coupling reaction between the two representative indeoindoles and a serial of aryl bromide under the above optimized conditions was executed. Some aryl iodides were also selected as electrophilic substrates where necessary. However, aryl chlorides could not receive the target products under the condition. The results were summarized in Tables 2 and 3.
|
|
Table 2 Substrate scope.a |
|
|
Table 3 Substrate scope.a |

Generally, both aryl bromides and iodides are substrates suitable for this reaction, and the iodides performed slightly better than their bromide congeners (Table 2 entry 1 vs. 2, entry 4 vs. 5, entry 10 vs. 11, entry 12 vs. 13). Concerning the aryl bromide, both their electronic and steric effect have considerable impacts on the coupling reaction. The electron-neutral aryl bromide were smoothly coupled with the indenoindole to give moderate to excellent yields (Table 2, entries 1, 3, 9 and 10, Table 3, entries 3 and 5), electron-deficient aryl bromide also delivered good product yields (entries 12 in Table 2 and entry 6 in Table 3). Interestingly, the nitrile did not play a role in activating the electrophilic substrate as usual, and instead, almost no product was afforded (Table 2, entry 7), presumably due to the strong coordinating ability of the p-CN, while those bearing strong electron-withdrawing groups seemed difficult to undergo the crossing-coupling desired. For example, 4-nitrobromobenzene and 4-bromobenzene carboxylic acid (unlisted in Table 2) did not react at all. Electrondonating groups gave a relatively low yield of the desired product (Table 2, entry 6). Furthermore, those containing the carbonyl group, such as 4-bromobenzaldehyde (Table 2, entry 8) provided no desired product, although the starting bromide was found to be consumed completely. This might be mainly due to the carbonyl group being capable of capturing the nucleophile and leading to a failure of the reduction of the Ni (Ⅱ) into the catalytically active low-valent species. The reaction is sensitive to the steric hindrance of the aryl bromide. Thus the reaction of 2-bromotoluene and 3, 4-dimethylbromobenzene with indenoindole gave low yield of 35% and=%, respectively (Table 2, entries 4 and 9), and orthosubstituted aryl bromides were coupled with indenoindole (entry 4 in Table 2 and entry 4 in Table 3) in relatively low yields. The main reason is presumably that the ortho-substituted aryl bromides were much more spatially congested to coordinate to the nickel center which already bears a bulky indenoindole amide species.
Furthermore, it is found that the indenoindole/heloarene coupling reaction is quite different from most of the nickelcatalyzed aromatic C--N coupling reported previously [20-22]. From the preliminary experimental observations, it is suggested that the catalytically active species might involve the Ni (Ⅰ) phosphine complexes in this reaction, which is consistent with our results: (1) very small amounts of biaryl by-products were detected in almost all cases; (2) chloroarenes were inert in the reaction, the conversion efficiency increases with the increasing of bromoarene-/iodoarenes concentration and the iodoarenes have higher reactivity; (3) the reaction proceeded only in the presence of strong bases with reducing ability such as butyl lithium. Arylnikel (Ⅱ) halide complexes are readily available compounds [26]. To discern the mechanism of the reaction, we conducted a stoichiometric reaction of indenoindole lithium with Ni (Ⅱ)-(a-aryl) complexes which may be regarded as theoxidative adducts of haloarenes to the nickel (0) as shown in Eq. (1). As a result, no N-aryl substituted indenoindole was received. This experiment indicated that the arylnickel (Ⅱ) complex is not the potential intermediate on the catalytic pathway. Thus, it should be reasonable to rule out the mechanism to follow an usual Ni (0)-Ni (Ⅱ) cycle for this nickel-catalyzed N-aryl substituted indenoindole derivatives synthesis.

|
(1) |
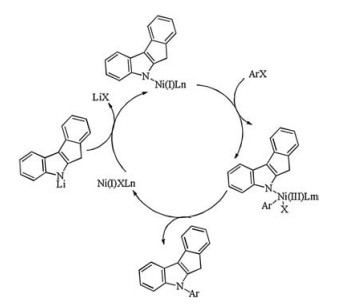
It was presumed that the catalytically active species might involve the Ni (Ⅰ) species in this reaction. Kochi and Tsou [27] has carefully investigated the oxidative addition of the nickel (0) on aryl halidesand proposed a route to in-situ forming the Ni (Ⅰ) species via a one-electron transfer of the in situ-generated Ni (0) species to the haloarene. Based on their studies, we proposed a route to generating the nickel (Ⅰ) intermediate as shown in Scheme 1. Firstly, the nickel (Ⅱ) compound was reduced to the nickel (0) species by butyl lithium agents, then the nickel (Ⅰ) species were formed via a single-electron transfer from nickel (0) to an aryl halide and the crossing reaction of the indenoindole derivatives with bromo-/iodoarenes was initiated.

|
Download:
|
| Scheme 1. A plausible route to in-situ generating the nickel (Ⅰ) species. | |
{kind=link}
Based on our experimental investigation, and combining previous studies [19, 28]. We propose a plausible mechanism that might follow a catalytic cycle of the Ni (Ⅰ)-Ni (Ⅲ) shuttle involving sequential oxidative addition, transmetallation and reductive elimination as shown in Scheme 2. For this nickel-catalyzed synthesis, efficient production of the nickel (Ⅰ) species would be the key step for the coupling reaction, and we are currently more inclined to consider the oxidative addition as the rate determining step. This conclusion may partly explain the reasons why aryl chlorides do not react under the condition and there always existed a little biaryl by-product.

|
Download:
|
| Scheme 2. A proposed mechanism for the arylation of indenoindole with bromo-/ iodoarenes catalyzed by NiCl2(PPh3)2. | |
{kind=link}
3. Conclusion
In summary, we have for the first time demonstrated the feasibility for Ni-catalyzed arylation of indenoindoles with bromo-/iodoarenes. A simple and practical method was provided for the synthesis of N-aryl substituted indenoindole. A preliminary investigation suggested that the mechanism of this reaction might be different from that of the normal Ni (0)-Ni (Ⅱ) shuttle. Further work to expand the scope of substrates and elucidate the mechanistic details is ongoing in our laboratory.
4. ExperimentalAll the chemicals were purchased commercially and used without any further treatment, unless otherwise stated. 1H NMR spectra were recorded on a BRUKER AVANCE 400 spectrometer. Mass spectra were obtained on a Bruker Daltonics Inc. APEXⅡ FTICR. Melting points were measured with an X-4 micro meltingpoint apparatus and uncorrected. All reactions were carried out under nitrogen atmosphere with oven-dried glassware and heated in an oil bath. Dioxane and toluene were distilled from sodium/ benzophenone before use. Grignard reagents and n-butyl lithium were purchased from commercial sources. All amines and haloarenes were available commercially and used without further purification. Column chromatography was performed on silica gel (200-300 mesh). All yields refer to isolated yields (average of two run) of compounds estimated to be >95% pure as determined by 1H NMR. The known compounds were partly characterized by melting points, MS, 1H NMR, and compared to authentic samples or the literature data.
Typical procedure for the N-aryl substituted indenoindole synthesis: An oven-dried 50 mL three-necked flask was charged with NiCl2(PPh3)2 (5 mol% relative to the indenoindole lithium, 33 mg) and 1 mmol indenoindole lithium. The flask was evacuated and backfilled with nitrogen, with the operation being repeated twice. Dried toluene (10 mL) was added via syringe. The resulted mixture was stirred for 5 min, a bromoarene (1.2 mmol) was added via syringe. The mixture was stirred for 10 min at room temperature and then heated at an oil bath of 85 -C for 12 h. The reaction mixture is allowed to cool to room temperature and added to 20 mL saturated ammonium chloride solution, then extracted with ethyl acetate (20 mL±3). The combined organic phases were evaporated under reduced pressure and the residue purified by column chromatography. The detail information about experimental procedures, the data of NMR and spectroscopy are deposited in Supporting information.
Date for representative examples are shown below:
5-Phenyl-5, 6-dihydroindeno[2, 1-b]indole (3aa): Colorless solid. 1H NMR (400 MHz, CDCl3): δ 7.92 (d, 1H, J=7.6 Hz), 7.69 (d, 1H, J=7.6 Hz), 7.57-7.52 (m, 5H), 7.43-7.35 (m, 3H), 7.30-7.21 (m, 4H), 7.11-7.05 (m, 2H), 3.80 (s, 2H). MS (EI): m/z 281 (M+).
5-Phenyl-5, 10-dihydroindeno[1, 2-b]indole (3ba): Pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.68 (d, 1H, J=8.0 Hz), 7.58-7.54 (m, 4H), 7.48 (t, 1H, J=3.2 Hz), 7.39 (d, 1H, J=6.4 Hz), 7.18-7.16 (m, 4H), 7.07 (d, 1H, J=7.2 Hz), 3.79 (s, 2H). MS (EI): m/z 281 (M+).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.11.002.
| [1] | C. Grandini, I. Camurati, S. Guidotti, N. Mascellani, L. Resconi, Heterocyclefused indenyl silyl amido dimethyl titanium complexes as catalysts for high molecular weight syndiotactic amorphous polypropylene. Organometallics 23 (2004) 344–360. |
| [2] | I.E. Nifant'ev, I.A. Kashulin, V.V. Bagrov, S.K. Abilev, Synthesis and study of the mutagenic activity of di (indeno[2, 1-b]indolyl)-and di (indeno[2, 1-b] pyrrolyl) methanes and -dimethylsilanes. Russ. Chem. Bull. Int. Ed. 50 (2001) 1439–1445. |
| [3] | Y.V. Kissin, L.A. Rishina, S.S. Lalavan, V.G. Krasheninnkikov, A new route to atactic polypropylene:the second life of premetallocene homogeneous polymerization catalyst. Polym. Chem. 53 (2015) 2124–2131. |
| [4] | H.H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger, R. Waymouth, Selfassembly of a ferromagnetically coupled manganese (Ⅱ) tetramer. Angew. Chem. Int. Ed. 34 (1995) 1143–1146. |
| [5] | J.A. Ewen, R.L. Jones, A. Razavi, J.D. Ferrara, Syndiospecific propylene polymerizations with group IVB metallocenes. J. Am. Chem. Soc. 110 (1988) 6255–6256. |
| [6] | A. Razavi, L. Peters, L.V. Nafpliotis, D.K. Den Dauw, J.L. Atwood, The geometry of the site and its relevance for chain migration and stereospecificity. Macromol. Symp. 89 (1995) 345–367. |
| [7] | P. Longo, A. Maricoda, L. D'Urso, M. Napoli, Syndiotactic-atactic stereoblock polystyrene obtained with ahapto-flexible catalyst. Macromolecules 47 (2014) 2214–2218. |
| [8] | L.E. Rosebrugh, V.M. Marx, B.K. Keitz, R.H. Grubbs, Highly active ruthenium metathesis catalysts exhibiting unprecedented activity and Z-selectivity. J. Am. Chem. Soc. 135 (2013) 10032–10035. |
| [9] |
(a) E. Brenner, Y. Fort, New efficient nickel (0) catalysed amination of aryl chlorides, Tetrahedron Lett. (391998) 5359-5362; (b) E. Brenner, R. Schneider, Y. Fort, Nickel-catalysed couplings of aryl chlorides with secondary amines and piperazines, Tetrahedron 55(1999) 12829-12842; (c) C. Desmarets, R. Schneider, Y. Fort, Nickel (0)/dihydroimidazol-2-ylidene complex catalyzed coupling of aryl chlorides and amines, J. Org. Chem. 67(2002) 3029-3036. |
| [10] | K. Matsubara, K. Ueno, Y. Koga, K. Hara, Nickel-NHC-catalyzed a-arylation of acyclic ketones and amination of haloarenes and unexpected preferential Narylation of 4-aminopropiophenone. J. Org. Chem. 72 (2007) 5069–5076. |
| [11] |
(a) B.H. Lipshutz, H. Ueada, Aromatic aminations by heterogeneous Ni0/C catalysis, Angew. Chem. Int. Ed. 39(2000) 4492-4494; (b) S. Tasler, B.H. Lipshutz, Nickel-on-charcoal-catalyzed aromatic aminations and kumada couplings:mechanistic and synthetic aspects, J. Org. Chem. 68(2003) 1190-1199. |
| [12] | G. Manolikakes, A. Gavryushin, P. Knochel, An efficient silane-promoted nickel-catalyzed amination of aryl and heteroaryl chlorides. J. Org. Chem. 73 (2008) 1429–1434. |
| [13] | M.J. Iglesias, A. Prieto, M.C. Nicasio, Well-defined allylnickel chloride/Nheterocyclic carbene[(NHC) Ni (allyl) Cl]complexes as highlyactive precatalysts for C-N and C-S cross-coupling reactions. Adv. Synth. Catal. 352 (2010) 1949–1954. |
| [14] | L. Ackermann, R. Sandmann, W.F. Song, Palladium-and nickel-catalyzed aminations of aryl imidazolylsulfonates and sulfamates. Org. Lett. 13 (2011) 1784–1786. |
| [15] | M. Tobisu, A. Yasutome, K. Yamakawa, T. Shimasaki, N. Chatani, Ni (0)/NHCcatalyzed amination of N-heteroaryl methyl ethers through the cleavage of carbon-oxygen bonds. Tetrahedron 68 (2012) 5157–5161. |
| [16] |
(a) F. Ullmannhem, Ueber eine neue bildungsweise von diphenylaminderivaten, Eur. J. Inorg. Chem. 36(1903) 2382-2384; (b) J.Lindley, Copperassistednucleophilicsubstitutionofarylhalogen, Tetrahedron 40(1984) 1433-1456. |
| [17] | A. Tlili, F. Monnier, M. Taillefer, Selective one-pot synthesis of symmetrical and unsymmetrical di-and triarylamines with a ligandless copper catalytic system. Chem. Commun. 48 (2012) 6408–6410. |
| [18] | N.S. Nandurkar, M.J. Bhanushali, M.D. Bhor, B.M. Bhanage, N-arylation of aliphatic, aromatic and heteroaromatic amines catalyzed by copper bis (2, 2, 6, 6-tetramethyl-3, 5-heptanedionate). Tetrahedron Lett. 48 (2007) 6573–6576. |
| [19] | S.V. Ley, A.W. Thomas, Modern synthetic methods for copper-mediated C (aryl)=O, C (aryl)=N, and C (aryl)=S bond formation. Angew. Chem. Int. Ed. 42 (2003) 5400–5449. |
| [20] | J.C. Antilla, A.S. Klapars, S.L. Buchwald, The copper-catalyzed N-arylation of indoles. J. Am. Chem. Soc. 124 (2002) 11684–11688. |
| [21] |
(a) C. Chen, L.M. Yang, Arylation of diarylamines catalyzed by Ni (Ⅱ)-PPh3 system, Org. Lett. 7(2005) 2209-2211; (b) C.Y. Gao, X. Cao, L.M. Yang, Nickel-catalyzed cross-coupling of diarylamines with haloarenes, Org. Biomol. Chem. 7(2009) 3922-3925. |
| [22] | C. Chen, L.M. Yang, Ni (Ⅱ)-(s-aryl) complex:a facile, efficient catalyst for nickel-catalyzed carbon-nitrogen coupling reactions. J. Org. Chem. 72 (2007) 6324–6327. |
| [23] | X.H. Fan, G. Li, L.M. Yang, Room-temperature nickel-catalyzed amination of heteroaryl/aryl chlorides with Ni (Ⅱ)-(s-aryl) complex as precatalyst. J. Organomet. Chem. 696 (2011) 2482–2484. |
| [24] | C.Y. Gao, L.M. Yang, Nickel-catalyzed amination of aryl tosylates. J. Org. Chem. 39 (2008) 1624–1627. |
| [25] | J.H. Huang, L.M. Yang, Nickel-catalyzed amination of aryl phosphates through cleaving aryl C-O bonds. Org. Lett. 13 (2011) 375–3750. |
| [26] |
(a) L. Cassar, S. Ferrara, M. Foá, Nickel-catalyzed cyanation of aromatic halides, Adv. Chem. Ser. 17(1974) 252-273; (b) J. van Soolingen, H.D. Verkruijsse, M.A. Keegstra, L. Brandsma, Nickelcatalyzed cyanation of 2-and 3-bromothiophene, Synth. Commun. 20(1990) 3153-3156; (c) L. Brandsma, S.F. Vasilevsky, H.D. Verkruijsse, Application of Transition Metal Catalysts in Organic Synthesis, Springer, New York, 1998, pp. 3-4. |
| [27] |
(a) T.T. Tsou, J.K. Kochi, Mechanism of oxidative addition reaction of nickel (0) complexes with aromatic halides, J. Am. Chem. Soc. 101(1979) 6319-6332; (b) J.K. Kochi, The role of electron transfer in organometallic chemistry, Pure Appl. Chem. 52(1980) 60-571. |
| [28] | M. Tobisu, T. Shimasaki, N. Chatani, Ni0-catalyzed direct amination of anisoles involving the cleavage of carbon-oxygen bonds. Chem. Lett. 38 (2009) 710–711. |