 2017, Vol. 28
2017, Vol. 28 



b College of Chemical and Environment Science, Shaanxi University of Technology, Hanzhong, 723001, China
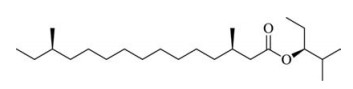
Nowadays, there is a sharper and sharper conflict between intense agricultural production and environmental issues. We all are facing a challenge caused by the detrimental effects resulting from traditional agricultural production. In fact, there has been a trend using some "green pesticide" in management of pest to suppress and control the pest population through mass trapping, attacticides and mating disruption [1]. In this context integrated pest management (IPM), the use of these low-dose substance would reduce the influence to the environment drastically [2]. Normally, the insect pheromone consists of some long-chained aliphatic compounds [3]. For example, the Paulownia bagworm, Cryptothelea variegata, is one of the most destructive forest defoliators widely distributed in Asia-Pacific countries, such as Japan, Korea, India, Indonesia, Thailand, New Zealand and in particular, China. This type of insect is quite difficult to control using common pesticides because its larvae often envelop themselves in a self-constructed bag. In 2006, Gries et al. firstly investigated the composition of the female-produced sex pheromone of C. variegata and identified (S)-2-methylpentan-3-yl-3, 13-dimethylpentadecanoate as a major component [4]. Moreover, the detailed chiral stereochemistry of C3 and C13 was ascertained after a field bioassay of four possible stereoisomers of 2-methylpentan-3-yl-3, 13-dimethylpentadecanoate as attractants to the male Paulownia bagworm [5]. Their forest trial revealed that (3R, 13R)-(S)-2-methylpentan-3-yl-3, 13-dimethylpentadecanoate (1, Fig. 1) exhibited the strongest luring activity to the male Paulownia bagworm [5]. Because of environmental issues, it is becoming increasingly unsustainable to kill pests using traditional, high-toxic pesticides. Obviously, using pheromones to control this kind of pest by luring and trapping would be a promising alternative. In 2010, Mori et al. reported the synthesis of all four of the stereoisomers of (S)-2-methylpentan-3-yl-3, 13-dimethylpentadecanoate starting from the expensive (R)-or (S)-2-methylbutan-1-ol and (R)-or (S)-citronellal using olefin cross metathesis as the key reaction [6a]. Recently, starting from the expensive (S)-propylene oxide, Taguri et al. reported syntheses of a set of four isomers of 1, through a sulfone mediated coupling process as the key step [6b]. As a continuation of our studies on the synthesis of insect pheromones [7], we have developed a practical route to synthesise this most active pheromone molecule. Herein, we report the efficient, cost-effective asymmetric synthesis of 1.

|
Download:
|
| Figure 1. Structure of the female Paulownia bagworm sex pheromone. | |
{kind=link}
2. Results and discussion
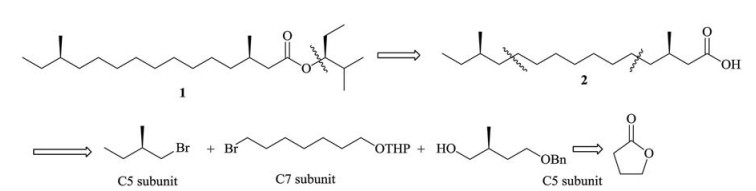
The retrosynthetic analysis is shown in Scheme 1. The target molecular was firstly disconnected from the chiral alcohol. Then, the main chain was dissembled into three parts, a chiral C5 subunit, a C7 subunit and another chiral C5 subunit. Thus, the focus became how to obtain the two chiral methyl subunits and realise the coupling reaction of Csp3--Csp3.

|
Download:
|
| Scheme 1. Retrosynthesis of 1. | |
{kind=link}
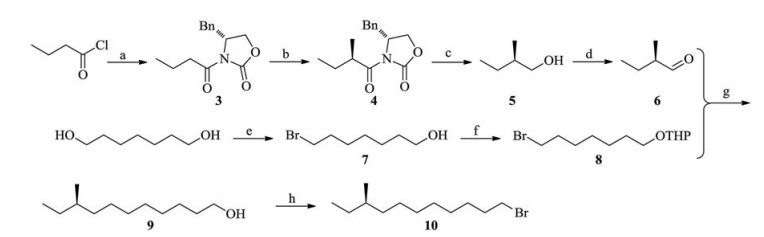
It is well known that chiral oxazolidin-2-one, namely Evans template, can form different steric space effects after protonation, which then give rise to an asymmetric alkylation process [8]. As shown in Scheme 2, the synthesis commenced from n-butyryl chloride, then the (R)-4-benzyloxazolidin-2-one was connected to give compound 3 in an excellent yield. After protonation by fresh prepared LDA and methylation with 3 equiv. of methyl iodide, the chiral induced compound 4 could be afforded in an 85% yield with the d.r. more than 99:1. Compound 4 was subjected to reduction in the presence of lithium borohydride to give chiral alcohol 5 in 93% yield ([α]D25 5.9 (c 1.12, CHCl3), lit. [9] [α] 5.8 (c 1.09, CHCl3)). Compound 5 could be transformed into compound 6 after PCC oxidation protocol. Thus, the first C5 subunit was obtained. The middle C7 subunit was prepared from 1, 7-heptanediole. It could be transformed into 7-bromo-heptanol selectively in conditions of 40% hydrobromic acid in the presence of catalytic iodine with a Dean-Stark trap. In order to couple with compound 6, the residual hydroxyl group was allowed to be protected by THP (tetrahydropyranyl) ether. Compound 8 was converted to its phosphonium salt with PPh3 in xylene at reflux. The Wittig reaction was allowed to connect two parts in THF, and then the coupled compound was hydrogenated to give 9 in 67% yield in three steps. After a conventional acidic procedure to remove THP ether, the obtained alcohol 9 was transformed into bromide 10 by the combination of CBr4andtriphenylphosphinein87% yield, [α]D25-2.9 (c 2.6, CHCl3).

|
Download:
|
| Scheme 2. Synthesis of C5 + C7 intermediate. Regents and reactants: (a) n-BuLi, (R)-4-benzyloxazolidin-2-one, dry THF, -78 ℃ to room temperature, overnight, 91%; (b) LDA, MeI, THF, -78 ℃ to room temperature, overnight, 85%; (c) LiBH4, THF, 0 ℃ to room temperature, 24 h, 93%; (d) PCC, CH2Cl2, 0 ℃ to room temperature, overnight, 82%; (e) 40% HBr, I2, toluene, 120 ℃, 24 h, 77%; (f) TsOH, DHP, CH2Cl2, 0 ℃ to room temperature, overnight, 84%; (g) ⅰ) PPh3, xylene, reflux 8 h; ⅱ) BuLi, -78 ℃, dry THF, overnight; ⅲ) Pd/C, H2, EtOAc, ⅳ) TsOH, CH3OH, H2O, 74% for 4 steps; (h) CBr4, Ph3P, CH2Cl2, 87%. | |
{kind=link}
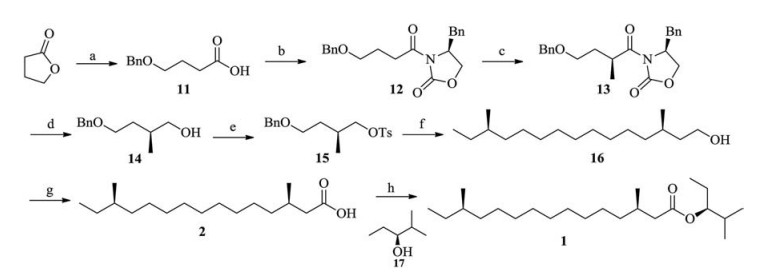
With the C5 + C7 intermediate 10 successfully synthesised, we next turned our attention to another C5 subunit, which was prepared from γ-lactone. As shown in Scheme 3, the ring-opening and benzylation in one-pot led to the efficient production of compound 11 on a large scale. The connection of compound 11 with (S)-4-benzyloxazolidin-2-one through activated mixed-anhydride with ethyl chloroformate at -78 ℃ for 6 h was accomplished in an 87% yield. The subsequent asymmetric methylation delivered compound 13 using sodium bis (trimethylsilyl) amide as a base with only one desirable diastereomer 14 in a 70% yield and a ratio of d.r. > 90:1. In the methylation step, the optimal conditions involved the slow addition of three equivalents of methyl iodide in diluted THF (tetrahydrofuran) solution over 2 h to avoid the dimethylated by-product. Removal of the auxiliary moiety by lithium aluminium hydride in dry THF and subsequent tosylation produced compound 15 in an excellent yield. As to the reduction and removal of the Evans auxiliary, the yield of the expensive lithium borohydride was higher than of lithium aluminium hydride. After the same coupling condition in the presence of Li2CuCl4 in the mixture of THF and NMP, compound 16 was produced. Removal of the benzyl group was accomplished through Pd (OH)2/C catalytic hydrogenation and the obtained alcohol was oxidised by Jones' reagent to afford acid 2 in a two-step 63% yield, The chiral alcohol 17 could be prepared through a reported kinetic resolution process [4]. After activation with trifluoroacetic anhydride, the total synthesis of target molecule 1, [α]D25 -3.8 (c 0.23, CHCl3) was finally achieved in a 75% yield.

|
Download:
|
| Scheme 3. Synthesis of 1. Regents and reactants: (a) BnCl, NaOH, reflux, 6 h, 87%; (b) ⅰ) EtOCOCl, Et3N, dry Et2O, 0 ℃ to room temperature; ⅱ) n-BuLi, (S)-4-benzyloxazolidin-2-one, dry THF, -78 ℃ to room temperature, overnight, 87%; (c) NaHMDS, MeI, dry THF, -78 ℃ to room temperature, overnight, 66%; (d) LiAlH4, dry THF, 0 ℃ to room temperature, 24 h, 83%; (e) TsCl, Et3N, DMAP, CH2Cl2, 0 ℃ to room temperature, overnight, 76%; (f) ⅰ) Compound 10, Mg, I2, dry THF, 80 ℃; ⅱ) Li2CuCl4, NMP, THF, 80 ℃, 87%; ⅲ) Pd (OH)2/C, H2, CH3OH, quantitatively; (g) CrO3, CH3COOH, room temperature, 24 h, 63%; (h) (CF3CO)2O, CH2Cl2, room temperature, 75%. | |
{kind=link}
3. Conclusions
We have successfully synthesised the most active stereoisomers of (3R, 13R)-(S)-2-methylpentan-3-yl-3, 13-dimethylpentadecanoate, the major and most active component of the sex pheromone of C. variegata. Evans template induction was adopted to produce the chiral methyl group moieties. Li2CuCl4 was used to catalyse the Csp3--Csp3 coupling, which was a key method in construction of the skeleton. Based on our synthetic route, a set of four diastereoisomers could be prepared with ease using only different Evans auxiliaries. Our results are highly beneficial for preventing and controlling Paulownia bagworm, C. variegata, in an economical and environmentally benign way.
4. ExperimentalAll anhydrous solvents and reagents were prepared from reagent grade materials using conventional methods. The reactions of air and moisture sensitive materials were carried out in flame dried glassware under a nitrogen atmosphere. The transfer of air and water sensitive solutions was completed using hypodermic syringes. The NMR data were recorded in CDCl3 solution with Bruker AC-500 or AM-400 MHz spectrometers, if not stated otherwise. The chemical shifts are reported in ppm relative to TMS. Column chromatography was generally performed on a silica gel (200-300 mesh) gradient elution with petroleum ether and ethyl acetate, and TLC inspections on silica gel GF-254 plates with petroleum ether:ethyl acetate (8:1, v/v), if not stated otherwise. The purities of the synthesised compounds were estimated by GC-MS using a Trace GC-MS 2000.
4.1. (R)-4-Benzyl-3-butyryloxazolidin-2-one 3In argon, to a cooled (-78 ℃) solution of (R)-4-benzyloxazolidin-2-one (21.6 g, 0.121 mol) in anhydrous THF (300 mL) was added n-BuLi (=mL, 2.5 M in hexane), and then the reaction mixture was stirred for 30 min. Butyryl chloride (14 mL) was slowly added at this temperature and the reaction mixture was stirred for 1 h. The reaction mixture was warmed to room temperature and maintained for another 8 h. Then, the mixture was quenched by saturated ammonia chloride and extracted by ethyl acetate. The organic phase was combined and washed with saturated brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography to produce 3 (27.4 g, 91%) as a colourless oil. [α]D27 -54.5 (c 3.00, CHCl3). 1H NMR (400 MHz, CDCl3): δ 1.01 (t, 3H, J=7.4 Hz), 1.68-1.77 (m, 2H), 2.76 (dd, 1H, J=13.4, 9.3 Hz), 2.83-3.00 (m, 2H), 3.30 (dd, 1H, J=13.3, 3.1 Hz), 4.15-4.22 (m, 2H), 4.64-4.70 (m, 1H), 7.20-7.35 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 173.2, 153.4, 135.3, 129.4, 128.9, 127.3, 66.1, =.1, 37.9, 37.3, 17.6, 13.6.
4.2. (R)-4-Benzyl-3-((R)-2-methylbutanoyl) oxazolidin-2-one 4To a stirred solution of 3 (3.5 g, 14.15 mmol) in anhydrous THF (20 mL) at-78 ℃, LDA (2.0 mol/L in THF, 8.88 mL, 17.75 mmol) was added dropwise under an argon atmosphere. After stirring for 1 h at -78 ℃, MeI (2.65 mL, 42.=mmol) was added and the reaction mixture was stirred for an additional 1 h at -78 ℃ and then warmed to room temperature. Upon completion of the reaction (monitored by TLC), the reaction mixture was quenched with saturated NH4Cl (10 mL) and extracted with EtOAc (70 mL±3). The combined organic extracts were washed with brine (50 mL), dried with anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography to afford 4 (3.2 g, 85%) as a colourless liquid. 1H NMR (400 MHz, CDCl3): δ 0.93 (t, 3H, J=7.4 Hz), 1.22 (d, 3H, J=7 Hz), 1.43-1.50 (m, 1H), 1.74-1.81 (m, 1H), 2.76 (dd, 1H, J=14.3, 9.8 Hz), 3.27 (dd, 1H, J=12.3, 3.1 Hz), 3.61-3.66 (m, 1H), 4.15-4.22 (m, 2H), 4.65-4.71 (m, 1H), 7.20-7.35 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 177.1, 153.0, 135.3, 129.4, 128.8, 127.3, 65.9, =.3, 39.1, 37.8, 26.3, 16.8, 11.6.
4.3. (R)-2-Methylbutanal 6To a solution of compound 4 (4.9 g, 18.8 mmol) in THF (120 mL) at 0 ℃, was added an aqueous solution of LiBH4 (0.18 g, 7.=mmol) portion-wise. The solution then was allowed to warm to room temperature naturally. After completion, the reaction mixture was neutralised by diluted HCl and extracted with EtOAc (50 mL±3). The combined organic extract was washed with brine (50 mL), dried with Na2SO4 and concentrated under atmospheric pressure. The crude product was purified by column chromatography to afford 5 (1.34 g, 83%) as a colourless liquid. 1H NMR (300 MHz, CDCl3): δ 0.91 (t, 3H, J=7.4 Hz), 1.22 (d, 3H, J=7.1 Hz), 1.20-1.35 (m, 1H), 1.39-1.56 (m, 1H), 1.67-1.77 (m, 1H), 3.29-3.45 (m, 2H). To a solution of compound 5 (1.2 g, 13.6 mmol) in CH2Cl2 (60 mL) at 0 ℃, was added PCC (pyridinium chlorochromate, 3.5 g, 16.3 mmol) portion-wise. The mixture was stirred at room temperature until completion. Then, ether was added to the system and filtered through a pad of silica gel. The filtrate was washed with diethyl ether, then was dried with Na2SO4 and concentrated in vacuo. The resultant compound 6 was used as obtained without further purification.
Compound 8 was prepared according to the literature [10] and the corresponding triphenyl phosphonium salt was also prepared. To a cooled solution of the salt (6.4 g, 11.7 mmol) in THF (80 mL) was added BuLi (12 mmol), and then maintained for 1 h. The resultant mixture was cooled to -78 ℃ again and the crude compound 6 (0.85 g, 10 mmol) in THF (15 mL) was added slowly. Through conventional workup and purification via column chromatography, the olefin product of the Wittig reaction was obtained. The crude produce was dissolved into methanol (50 mL) and 4-methylbenzenesulfonic acid (200 mg) was added. The reaction mixture was stirred overnight and then 20%Pd (OH)2/C (500 mg) was added to catalyse the hydrogenation process. After evaporation, the residue was purified through column chromatography to afford chiral alcohol 9 (1.4 g, 74%) as colourless oil. IR (neat, cm-1): 2956, 2928, 28=, 1454, 1100, 734, 697. 1H NMR (400 MHz, CDCl3): δ 0.84 (t, 3H, J=7.4 Hz), 0.95 (d, 3H, J=6.7 Hz), 1.16-1.23 (m, 1H), 1.28-1.39 (m, 7H), 1.59-1.66 (m, 2H), 2.00-2.05 (m, 2H), 2.31-2.34 (m, 1H), 3.45-3.49 (m, 2H), 5.08-5.13 (m, 1H), 5.28-5.34 (m, 1H), 7.26-7.35 (m, 5H). 13C NMR (100 MHz, CDCl3): 138.6, 136.1, 128.4, 128.3, 127.6, 127.4, 72.8, 70.4, 33.3, 30.2, 29.8, 29.7, 29.1, 27.4, 26.0, 21.0, 11.9. HRESIMS (C19H30O): 274.2710.
4.5. (R)-1-Bromo-9-methylundecane 10At 0 ℃, to a solution of compound 9 (5.0 g, 27.2 mmol) in dichloromethane (150 mL) was added CBr4 (11.3 g, 34 mmol) and triphenylphosphine (11.4 g, 44 mmol). Then, the ice bath was removed and the reaction mixture was stirred for another 5 h. The reaction mixture was evaporated and the residue was purified through column chromatography to afford chiral bromide 10 as colourless oil (5.88 g, 87%). [α]D23 3.2 (c 0.61, CHCl3). IR (neat, cm-1): 2959, 2925, 2854, 1462. 1H NMR (400 MHz, CDCl3): δ 0.83-0.87 (m, 6H), 1.06-1.31 (m, 2H), 1.27-1.34 (m, 11H), 1.40-1.44 (m, 2H), 1.82-1.89 (m, 2H), 3.41 (t, 2H, J=7.0 Hz). 13C NMR (100 MHz, CDCl3): 36.5, 34.3, 34.0, 32.8, 29.8, 29.4, 28.7, 28.1, 27.0, 19.2, 11.4.
4.6. (3R, 13R)-3, 13-Dimethylpentadecan-1-ol 16Compound 15 was prepared through a similar literature procedure to 6 and the yield is shown in Scheme 3. At 0 ℃, to a flask charged with 14 (2.88 g, 20 mmol) in redistilled pyridine (30 mL) was added 4-methylbenzene-1-sulfonyl chloride (4.2 g, 22 mmol) potion-wise. The reaction mixture was stirred for 2 h and poured into diluted HCl, and then extracted with ethyl acetate. All the organic phases were combined and washed with water, brine and 5% CuSO4, successively. The obtained compound 15 was used directly without further purification. Under argon, to another three-necked flask was charged Mg (200 mg, 8.7 mmol) and the chiral compound 10 (1.8 g, 7.25 mmol) was introduced slowly, thus, a Grignard reagent of compound 10 can be obtained successfully. Dilithium tetrachlorocuprate (II) solution (7 mL, 0.1 mol/L in THF, from Sigma-Aldrich) and anhydrous NMP (3 mL) were added into the above solution. Compound 15 was then introduced into the reaction mixture through a double-tipped needle. Thereafter, the reaction mixture was refluxed for 10 h and evaporated to dryness, purified through a column to give (3R, 13R)-3, 13-dimethylpentadecyl benzyl ether. This ether was dissolved in methanol (40 mL) and hydrogenated in the presence of catalytic Pd (OH)2 to give 16 (1.58 g, 87%). [α]D23 -1.4 (c 0.54, CHCl3). IR (neat, cm-1): 3332, 2958, 2924, 2853, 1463, 1377, 1057, 735. 1H NMR (400 MHz, CDCl3): d 0.82-0.89 (m, 9H), 1.07-1.16 (m, 2H), 1.16-1.41 (m, 20H), 1.50-1.64 (m, 2H), 3.62-3.72 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 61.2, 39.9, 37.1, 36.6, 34.4, 30.0, 29.9, 29.7, 29.4, 27.1, 26.9, 19.6, 19.2, 11.4.
4.7. (3R, 13R)-3, 13-Dimethylpentadecanoic acid 2To a solution of CrO3 (1 g, 10 mmol) in 80% acetic acid (10 mL) was added dropwise a solution of compound 16 (720 mg, 2.8 mmol) in acetic acid (7 mL). The reaction mixture was allowed to stir at room temperature for 24 h. Upon completion, it was diluted with water (30 mL) and extracted with ether (30 mL±5). The combined extracts were treated with a conventional workup and purified through column chromatography to give 2 (600 mg, 80%). [α]D24 -1.5 (c 1.25, CHCl3). IR (neat, cm-1): 3500-3200, 2959, 2925, 2854, 1709, 1363. 1H NMR (400 MHz, CDCl3): δ 0.83-0.89 (m, 6H), 0.96 (d, 3H, J=6.7 Hz), 1.06-1.16 (m, 2H), 1.16-1.38 (m, 20H), 2.14 (dd, 1H, J=14.9, 8.2 Hz), 2.35 (dd, 1H, J=14.9, 5.9 Hz). 13C NMR (100 MHz, CDCl3): δ 178.2 41.6, 36.6, 34.3, 30.1, 30.0, 29.7, 29.6, 29.5, 27.1, 26.8, 19.6, 19.2, 11.4.
4.8. (3R, 13R)-(S)-2-Methylpentan-3-yl-3, 13-dimethylpentadecanoate 1To a solution of compound 2 (50 mg, 0.2 mmol) in dichloromethane (20 mL) was added two drops of trifluoroacetic anhydride. Compound 17 (25 mg, 0.25 mmol) in dichloromethane (1 mL) was added after 30 min. The reaction mixture was evaporated and purified through column chromatography to give 1 (35 mg, 75%). 1H NMR (400 MHz, CDCl3): δ 0.83-0.94 (m, 18H), 1.04-1.38 (m, 21H), 1.48-1.58 (m, 2H), 1.80-1.85 (m, 1H), 2.11 (dd, 1H, J=14.5, 8.2 Hz), 2.31 (dd, 1H, J=14.5, 5.9 Hz), 4.66-4.70 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 173.3, 79.4, 42.2, 36.7, 36.6, 34.3, 30.8, 30.3, 30.0, 29.7, 29.6, 29.4, 27.1, 26.9, 23.9, 19.7, 19.2, 18.5, 17.6, 11.4, 9.8.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (No. 31301712) is greatly appreciated. Zhenting Du would like to thank the Opening Funds of the Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences for partial financial support. Zhenting Du is deeply indebted to Hong-Li Zhang for his assistance in acquiring NMR spectra.
| [1] |
(a) P. Trematerra, Advances in the use of pheromones for stored-product protection, J. Pest Sci. 85(2012) 285-299; (b) M.A.Silva, G.C.D. Bezerra-Silva, T. Mastrangelo, The host marking pheromone application on the management of fruit flies-a review, Braz. Arch. Biol. Technol. 55(2012) 835-842. |
| [2] | G.Z. Cui, J.J. Zhu, Pheromone-based pest management in China:past, present, and future prospects. J. Chem. Ecol. 42 (2016) 557–570. DOI:10.1007/s10886-016-0731-x |
| [3] |
(a) C. Che, Z.N. Zhang, A facile synthetic method for (3Z 6Z, 9S, 10R)-9, 10-epoxy-3, 6-heneicosadiene, sex pheromone component of Hyphantria cunea (drug), Chin. Chem. Lett. 16(2005) 468-470; (b) B. Sun, L.Z. Peng, X.S. Chen, Y.L. Li, Y. Li, Short and stereoselective synthesis of the (-)-(5R 6S)-6-acetoxyhexadecane-5-olide, Chin. Chem. Lett. 15(2004) 1177-1178. |
| [4] | R. Gries, G. Khaskin, Z.X. Tan, (1S)-1-Ethyl-2-methylpropyl 3, 13-dimethylpentadecanoate:major sex pheromone component of Paulownia bagworm Clania variegata. J. Chem. Ecol. 32 (2006) 1673–1685. DOI:10.1007/s10886-006-9101-4 |
| [5] | B.G. Zhao, A.M. El-Sayed, S.F. Wang, Determination of the sex hormone 3-D configuration from the Clania variegata by field trapping. China Forest. Sci. Technol. 23 (2009) 21–22. |
| [6] |
(a) K. Mori, T. Tashiro, B. Zhao, D.M. Suckling, A.M. El-Sayed, Pheromone synthesis. Part 243:synthesis and biological evaluation of (3R, 13R, 1'S)-1'-ethyl-2'-methylpropyl 3, 13-dimethyl pentadecanoate, the major component of the sex pheromone of Paulownia bagworm, Clania variegata, and its stereoisomers, Tetrahedron 66(2010) 2642-2653; (b) T. Taguri, M. Yamamoto, T. Fujii, Y. Muraki, T. Ando, Synthesis of four stereoisomers of (S)-2-methylpent-3-yl 3, 13-dimethylpentadecanoate, a sex pheromone of the bagworm moth Clania variegate, using stereospecific inversion of secondary sulfonates as a key step, Eur. J. Org. Chem. (2013) 6924-6933. |
| [7] |
(a) T. Zhang, J. Feng, C. Cai, X. Zhang, Synthesis and field test of three candidates for Soybean Pod Borer's sex pheromone, Nat. Prod. Commun. 9(2011) 1323-1326; (b) T. Zhang, W.L. Ma, T.R. Li, et al., A facile asymmetric synthesis of (S)-14-methyl-1-octadecene, the sex pheromone of the peach leafminer moth, Molecules 18(2013) 5201-5208. |
| [8] |
(a) D.A. Evans, M.D. Ennis, D.J. Mathre, Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of α-substituted carboxylic acid derivatives, J. Am. Chem. Soc. 104(1982) 1737-1739; (b) J.S. Yadav, N.N. Yadav, T.S. Rao, B.V.S. Reddy, A.A.K.A. Ghamdi, Enantioselective total synthesis of (+)-vittatalactone, Eur. J. Org. Chem. (2011) 4603-4608. |
| [9] | M.G. Organ, Y.V. Bilokin, S. Bratovanov, Approach toward the total synthesis of orevactaene:convergent and stereoselective synthesis of the C18-C31 domain of orevactaene. Evidence for the relative configuration of the side chain. J. Org. Chem. 67 (2002) 5176–5183. DOI:10.1021/jo0201777 |
| [10] |
(a) S. Shan, Y. Jun, Studies on insect pheromones. I. Stereoselective synthesis of the sex pheromone of Khapra beetle, (Z)-14-methyl-8-hexadecenal, Acta Chim. Sinica 46(1988) 1045-1048; (b) E.G. Neeland, J.P. Ounsworth, R.J. Sims, L. Weiler, Synthesis, conformational analysis, and stereoselective reduction of 14-membered ring 3-keto lactones, J. Org. Chem. 59(1994) 7383-7394. |