 2017, Vol. 28
2017, Vol. 28 



b Chongqing Engineering Laboratory of Targeted and Innovative Therapeutics, Chongqing Key Laboratory of Kinase Modulators as Innovative Medicine, IATTI, Chongqing University of Arts and Sciences, Chongqing 402160, China;
c Department of Oncology, Chengdu Military General Hospital, Chengdu 610083, China
Nitrogen-containing heterocycles are important compounds that can be found in a wide range of drugs and biologically relevant molecules [1]. For this reason, research towards the development of novel and efficient strategies for the construction of these compounds represents one of the most active areas of present day synthetic organic chemistry [2]. Multicomponent reactions (MCRs) have become useful tools for the diversity-oriented synthesis of complex heterocyclic compounds [3]. One of the powerful multicomponent reactions is the Ugi reaction, which involves the effective multicomponent reaction of an aldehyde (or ketone), amine, isonitrile and carboxylic acid to afford an α-acylamino carboxamide adduct [4, 5]. Tandem reaction sequences involving the use of an Ugi reaction, followed by various post-condensation transformations, represent extremely powerful synthetic methods for the construction of heterocyclic compounds with elaborate substitution patterns [6]. For example, Ugi/Buchwald-Hartwig/ Michael [7], Ugi/Heck [8], Ugi/Aldo [9], Ugi/Bischler Napieralski [10] and Ugi/gold-catalyzed [11] reaction sequences have been reported to provide efficient access to a wide variety of cyclic scaffolds.
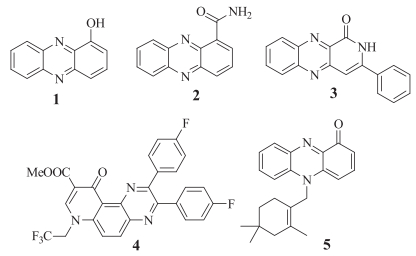
The quinoxaline skeleton is found in a wide range of biologically active natural products (1, 2, 5, Fig. 1) and synthetic compounds (3, 4, Fig. 1) [12]. Notably, quinoxaline derivatives have been reported to display a wide range of biological properties, including anticancer [13], antitumor (i.e., thymidylate synthase inhibitors) [14], antibacterial [15], antifungal [16], antihypertensive [17] and dopamine agonist [18] activities. In this study, we used a UDC, followed by an intermolecular nucleophilic substitution reaction to prepare a series of novel quinolino[3, 4-b]quinoxalin-6(5H)-one compounds. It is noteworthy, however, these compounds were formed unexpectedly during our efforts to prepare a related scaffold and therefore represent a serendipitous discovery.

|
Download:
|
| Figure 1. Quinoxaline natural and prepared products. | |
{kind=link}
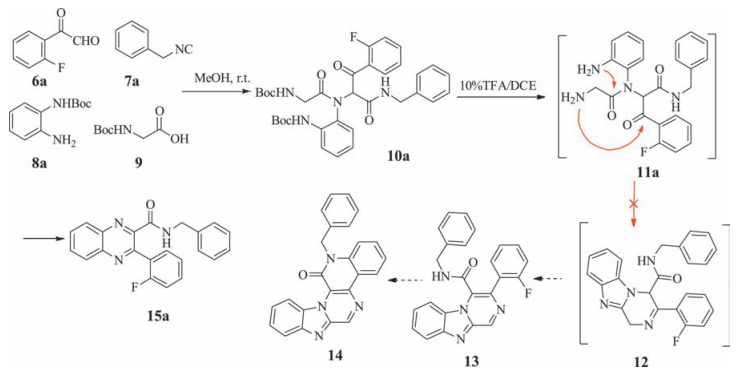
The synthesis of a series of fused piperazine-benzimidazoles via an UDC strategy was reported [19]. It was envisaged that we could expand the scope of this research to the construction of much more interesting and structurally complex compounds with a wide range of biological activities. During the last decade, G-quadruplexes have emerged as valid targets for the development of new anticancer drugs [20]. Numerous compounds have been reported to target these structures, and some of these compounds progressed into preclinical or clinical trials for the treatment of cancer. It is noteworthy that most of the compounds designed against G-quadruplexes are based on polycyclic heteroaromatic scaffolds, which could be constructed from Ugi products following a series of nucleophilic substitution reactions to provide access to additional aromatic ring structures. It was therefore envisioned that the reaction of 2-oxo-2-phenylacetaldehyde 6a with isonitrile 7a, N-Boc-protected-phenylenediamine 8a and N-Boc amino acetic acid 9 under the tandem UDC conditions would provide access to the corresponding benzo[4, 5]imidazo[1, 2-a]pyrazine derivative 14, as shown in Scheme 1.

|
Download:
|
| Scheme 1. Ugi/deprotection/cyclization (UDC) strategy. | |
{kind=link}
2. Results and discussion
The Ugi product 10a was obtained in good yield and subjected to a Boc-deprotection step to afford intermediate 11a. It was anticipated that compound 11a would react as indicated by the arrows shown in Scheme 1 to give compound 12. This material would then undergo a dehydrogenation reaction to give intermediate 13, followed by a nucleophilic aromatic substitution reaction to provide the target compound 14 in the presence of an inorganic base catalyst, such as Cs2CO3 or Na2CO3. In practice, however, the UDC product resulting from the reaction of 11a was determined to be the quinoxaline derivative 15a instead of the expected product 12. Notably, the formation of 15a involved the cleavage of the amino acid group from 11a during the UDC strategy when the cyclization was conducted under acidic condition. To develop a detailed understanding and further expand the scope of this process, we investigated the application of these reaction conditions to a series of different starting materials (Scheme 2).

|
Download:
|
| Scheme 2. Scope of the UDC strategy. | |
{kind=link}
The results of these reactions revealed that they gave rise to the corresponding quinoxaline products. The Ugi product 10 was obtained following the reaction of the N-Boc amino acetic acid 9 with N-Boc-protected-phenylenediamine 8, 2-oxo-2-phenylacetaldehyde 6 and isonitrile 7 in methanol at room temperature overnight, and used in the next reaction without purification. The methanol solvent was removed under a gentle stream of nitrogen to give a residue, which was dissolved in 10% TFA/DCE and heated under microwave irradiation to give the cyclized intermediate 16. Then, it underwent a deprotection step to give compounds 15a-g in 54%-72% yields over the two steps. All of these reactions provided access to the desired quinoxaline compounds via the UDC strategy. Although this strategy did not provide access to the desired compounds, it did provide an efficient method for the preparation of quinoxaline derivatives, following the modification of the reaction conditions. Overall, the outcome of this reaction can be attributed to the loss of the amino acid moiety (glycine) under the microwave irradiation conditions.
In addition, it is easy to consider an "acid-less" Ugi type reaction and phenylphosphonic acid (PPOA) is usually used as the catalyst [21-23]. We previously reported the intermolecular nucleophilic substitution reaction of an Ugi product as a strategy for the formation of compounds containing a new ring system via the formation of a C—N bond [24, 25]. Based on these reports, it was envisaged that the Ugi reaction products could also be used for the design and synthesis of several new scaffolds. To test this hypothesis, we conducted a series of acidless Ugi-type reactions according to the UDC strategy, followed by an intermolecular nucleophilic substitution reaction to provide access to a series of novel quinolino[3, 4-b]quinoxalin-6(5H)-one compounds. The products resulting from this new method are shown in Scheme 3.

|
Download:
|
| Scheme 3. Scope of this acid-free UDC strategy for the synthesis of quinolino[3, 4-b]quinoxalin-6(5H)-ones. | |
{kind=link}
A three-component Ugi-type reaction involving 7, 8 and 17 provided access to the corresponding quinoxaline compound 20 in two steps. A microwave temperature of 110 ℃ was required to affect the deprotection and cyclization steps of the UDC strategy. The results of these experiments revealed that the use of phenylphosphonic acid as a catalyst for the Ugi reaction provided satisfactory yields. The last step in this process (i.e., the nucleophilic substitution reaction), was performed with a series of different isonitriles, leading to the formation of the corresponding quinolino[3, 4-b]quinoxalin-6(5H)-ones 20a-f in 37%-54% yields over three steps. However, the use of tert-butyl isocyanide under the conditions required of the UDC strategy gave yields of 54% and 63% for compounds 15f and 15g, respectively. The nucleophilic substitution reactions of these two compounds were initially tested at a high temperature of 180 ℃ in dimethylformamide. Under these conditions, however, large amounts of the starting materials were recovered, with only trace quantities of the desired products being identified by LC/MS. Several bases, including K2CO3, Na2CO3 and NaOH, were also tested in the reaction, but failed to provide an increase in the product yields. The steric effect of the tert-butyl group was thought to be the main reason for these results. The use of a one-pot procedure did not require the conversion of a carboxylic acid, and was therefore more suitable for the synthesis of a large number of compounds [26].
3. ConclusionIn conclusion, we have synthesized a series of quinolino[3, 4-b]-quinoxalin-6(5H)-one derivatives using an Ugi/deprotection/cyclization (UDC) strategy followed a nucleophilic aromatic substitution reaction. This multicomponent reaction proceeded under acidless conditions to give the desired products in good yields and could be used in a variety of applications in medicinal chemistry.
4. ExperimentalAll reagents, unless otherwise stated, were used as received from commercial suppliers. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker Avance 400 spectrometer using CDCl3 or DMSO-d6 as solvent and TMS as internal standard (δ in ppm). Abbreviations used for NMR signals are: s=singlet, d=doublet, dd=doublet of doublets, dt=doublet of triplets, q=quartet, t=triplet, td=triplet of doublets and m=multiplet. LC/MS were recorded on a Shimadzu LCMS-2020. All microwave irradiation experiments were carried out in a Biotage® Initiator Classic microwave apparatus with continuous irradiation power from 0 to 400 W with utilization of the standard absorbance level of 250 W maximum power.
4.1. General procedures for the preparation of compounds 15a-gTo a magnetically stirred solution of an aldehyde (0.50 mmol) in MeOH (1.0 mL), an amine (0.50 mmol) was added in a 5 mL microwave vial. The solution was stirred for 10 min at room temperature. Then, an acid (0.50 mmol) and an isonitrile (0.50 mmol) were added separately. The mixture was stirred at room temperature overnight. The reaction was monitored by TLC and the solvent was removed under a nitrogen stream. The residue in the same vial was dissolved in 10% TFA/DCE (3.0 mL) and placed back in microwave and heated to 110 ℃ for 20 min. The microwave vial was then cooled to room temperature, the solvent removed under reduced pressure, the residue dissolved with EtOAc (15.0 mL), washed with saturated Na2CO3 and brine. The organic layer was dried over MgSO4 and concentrated. The residue was purified by silica gel column chromatography using a gradient of ethyl acetate/hexane (0-60%) to give the targeted compound 15a-g.
N-Benzyl-3-(2, 4-dichlorophenyl)-6, 7-dimethylquinoxaline-2-carboxamide (15a): Yellow solid, yield 67%. 1H NMR (400 MHz, CDCl3): δ 8.24 (t, 1H, J=5.5 Hz), 7.90 (d, 2H, J=22.6 Hz), 7.49-7.45 (m, 2H), 7.41 (dd, 1H, J=8.2, 1.9 Hz), 7.38-7.32 (m, 4H), 7.32-7.27 (m, 1H), 4.63 (d, 2H, J=5.9 Hz), 2.53 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 165.8, 160.6, 157.3, 153.2, 143.9, 140.6, 138.6, 131.8, 130.6, 129.7, 129.1, 129.0, 128.7, 128.3, 128.0, 127.4, 44.0, 20.5, 20.4. LC/MS calcd. for C24H20Cl2N3O, 436 [M+H]+; found 436.
N-Benzyl-3-phenylquinoxaline-2-carboxamide (15b): Yellow solid, yield 65%. 1H NMR (400 MHz, CDCl3): δ 8.18 (d, 1H, J=8.4 Hz), 8.10 (d, 1H, J=8.2 Hz), 7.88-7.76 (m, 3H), 7.73-7.67 (m, 2H), 7.52-7.46 (m, 3H), 7.37 (d, 3H, J=4.4 Hz), 7.34-7.28 (m, 1H), 4.66 (d, 2H, J=6.0 Hz). 13C NMR (100 MHz, CDCl3): δ 164.9, 153.8, 145.0, 142.6, 139.3, 138.6, 138.0, 131.7, 130.4, 129.4, 129.2, 128.9, 128.8, 128.2, 128.0, 127.6, 43.8. LC/MS calcd. for C22H18N3O, 340 [M +H]+; found 340.
N-Benzyl-6, 7-dimethyl-3-phenylquinoxaline-2-carboxamide (15c): Yellow solid, yield 72%. 1H NMR (400 MHz, CDCl3): δ 7.92 (s, 1H), 7.84 (s, 2H), 7.67 (dd, 2H, J=6.5, 3.1 Hz), 7.47 (dd, 3H, J=5.0, 1.7 Hz), 7.36 (d, 3H, J=4.3 Hz), 7.34-7.28 (m, 1H), 4.64 (d, 2H, J=6.0 Hz), 2.52 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 165.1, 152.9, 143.9, 142.8, 141.6, 141.3, 138.9, 138.3, 138.1, 128.9, 128.8, 128.3, 128.1, 128.0, 127.6, 43.8, 20.6, 20.4. LC/MS calcd. for C24H22N3O, 368 [M+H]+; found 368.
N-Benzyl-3, 6, 7-trimethylquinoxaline-2-carboxamide (15d): Yellow solid, yield 58%. 1H NMR (400 MHz, CDCl3): δ 8.40 (s, 1H), 7.77 (d, 2H, J=18.1 Hz), 7.42-7.29 (m, 5H), 4.70 (d, 2H, J=6.0 Hz), 3.13 (s, 3H), 2.50 (s, 3H), 2.47 (s, 3H). 13C NMR (100 MHz, CDCl3): d 164.9, 152.7, 145.0, 142.5, 142.0, 141.0, 140.4, 140.2, 139.9, 139.2, 138.3, 138.1, 128.8, 128.2, 128.0, 127.9, 127.8, 127.5, 43.6, 22.4, 20.4, 20.2. LC/MS calcd. for C19H20N3O, 306 [M+H]+; found 306.
N-Cyclohexyl-3, 6, 7-trimethylquinoxaline-2-carboxamide (15e): Yellow solid, yield 64%. 1H NMR (400 MHz, CDCl3): δ 7.92 (d, 1H, J=7.8 Hz), 7.79 (d, 2H, J=5.7 Hz), 4.04-3.92 (m, 1H), 3.10 (s, 3H), 2.49 (d, 6H, J=3.9 Hz), 2.06 (d, 2H, J=12.1 Hz), 1.79 (d, 2H, J=10.2 Hz), 1.53-1.31 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 164.0, 153.3, 142.7, 142.2, 141.9, 140.1, 138.1, 128.0, 127.5, 48.4, 33.1, 25.7, 25.0, 24.7, 20.5, 20.2. LC/MS calcd. for C18H24N3O [M+H]+, 298; found 298.
N-(tert-Butyl)-3-(2-fluorophenyl) quinoxaline-2-carboxamide (15f): Yellow solid, yield 63%. 1H NMR (400 MHz, CDCl3) δ 8.24-8.05 (m, 2H), 7.88-7.77 (m, 2H), 7.71 (td, 1H, J=7.5, 1.7 Hz), 7.56 (s, 1H), 7.35-7.31 (m, 1H), 7.20 (dd, 1H, J=10.9, 4.1 Hz), 7.11-6.99 (m, 1H), 1.51 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 164.0, 161.4, 159.0, 148.77, 146.2, 142.7, 139.3, 131.6, 131.0, 130.9, 130.8, 130.4, 129.3, 129.2, 127.6, 127.4, 124.5, 124.5, 115.1, 114.8, 51.7, 28.6. LC/MS calculated for C19H19FN3O [M+H]+, 324; found 324.
N-(tert-Butyl)-3-(2, 4-dichlorophenyl) quinoxaline-2-carboxamide (15g): Yellow solid, yield 54%. 1H NMR (400 MHz, CDCl3): δ 8.17 (dd, 2H, J=5.8, 4.0 Hz), 7.87 (dd, 2H, J=6.2, 3.1 Hz), 7.69 (s, 1H), 7.54 (d, 1H, J=8.1 Hz), 7.49-7.38 (m, 2H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 163.0, 151.0, 145.410, 142.5, 139.5, 137.6, 135.2, 133.4, 131.7, 131.0, 130.8, 129.4, 129.2, 128.9, 127.6, 51.5, 28.6. LC/MS calcd. for C19H18Cl2N3O, 374 [M+H]+; found 374.
4.2. General procedures for the preparation of compounds 20a-fTo a magnetically stirred solution of an aldehyde (0.50 mmol) in MeOH (1.0 mL), an amine (0.50 mmol) was added in a 5 mL microwave vial. The solution was stirred for 10 min at room temperature. Then, phenylphosphonic acid (0.05 mmol) and an isonitrile (0.50 mmol) were added separately. The mixture was stirred at room temperature overnight. The reaction was monitored by TLC and the solvent was removed under a nitrogen stream. The residue in the same vial was dissolved in 10% TFA/DCE (3.0 mL) and placed back in microwave and heated to 110 ℃ for 20 min. The solvent was removed under a nitrogen stream and diluted with DMF (3 mL) in the same vial. Cs2CO3 (2.0 mmol) was added and the vial was placed back in microwave and heated to 150 ℃ for 30 min. The microwave vial was then cooled to room temperature, the residue dissolved with EtOAc (15.0 mL), washed with brine. The organic layer was dried over MgSO4 and concentrated. The residue was purified by silica gel column chromatography using a gradient of ethyl acetate/hexane (20%-80%) to afford products 20a-f.
5-Benzylquinolino[3, 4-b]quinoxalin-6(5H)-one (20a): Yellow solid, yield 51%. 1HNMR (400 MHz, CDCl3): δ 9.02 (d, 1H, J=7.6 Hz), 8.48 (d, 1H, J=8.1 Hz), 8.27 (d, 1H, J=8.4 Hz), 7.92 (dt, 2H, J=14.7, 7.0 Hz), 7.53 (t, 1H, J=7.5 Hz), 7.45-7.27 (m, 7H), 5.73 (s, 2H). 13C NMR (100 MHz, CDCl3): δ 160.9, 145.1, 144.5, 143.1, 138.6, 136.0, 132.9, 132.4, 131.0, 130.7, 129.2, 128.9, 127.5, 126.8, 126.1, 123.5, 120.0, 115.9, 47.0. LC/MS calcd. for C22H16N3O, 338 [M+H]+; found 338.
5-Cyclohexylquinolino[3, 4-b]quinoxalin-6(5H)-one (20b): Yellow solid, yield 48%. 1H NMR (400 MHz, CDCl3): δ 9.02 (d, 1H, J=7.9 Hz), 8.41 (d, 1H, J=8.3 Hz), 8.23 (d, 1H, J=8.2 Hz), 7.94-7.78 (m, 2H), 7.63 (d, 2H, J=4.0 Hz), 7.42-7.33 (m, 1H), 4.86-4.35 (m, 1H), 2.82 (d, 2H, J=11.3 Hz), 2.04-1.78 (m, 6H), 1.48 (d, 2H, J=13.7 Hz). 13C NMR (100 MHz, CDCl3): δ 160.7, 144.9, 144.4, 143.0, 139.3, 132.5, 132.0, 130.9, 130.5, 129.1, 126.4, 123.0, 120.4, 115.4, 58.8, 28.9, 26.7, 25.5. LC/MS calcd. for C21H20N3O [M+H]+, 330; found 330.
5-(2, 6-Dimethylphenyl) quinolino[3, 4-b]quinoxalin-6(5H)-one (20c): Yellow solid, yield 44%. 1H NMR (400 MHz, CDCl3): δ 9.08 (dd, 1H, J=7.7, 1.5 Hz), 8.44 (d, 1H, J=8.4 Hz), 8.38-8.24 (m, 1H), 8.04-7.83 (m, 2H), 7.49-7.30 (m, 4H), 7.15-7.01 (m, 1H), 6.60 (d, 1H, J=8.0 Hz), 2.07 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 159.3, 145.5, 144.6, 143.0, 138.8, 136.1, 135.3, 133.0, 132.6, 131.1, 130.8, 129.3, 128.7, 128.2, 126.1, 123.8, 119.8, 115.5, 17.7. LC/MS calculated for C23H18N3O [M+H]+, 352; found 352.
3-Chloro-5-(2, 6-dimethylphenyl) quinolino[3, 4-b]quinoxalin-6 (5H)-one (20d): Yellow solid, yield 49%. 1H NMR (400 MHz, CDCl3): δ 9.01 (d, 1H, J=8.5 Hz), 8.50 (d, 1H, J=5.9 Hz), 8.30 (d, 1H, J=8.5 Hz), 8.01-7.88 (m, 2H), 7.48-7.28 (m, 4H), 6.59 (d, 1H, J=1.3 Hz), 2.08 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 159.3, 144.8, 143.0, 139.7, 138.8, 135.9, 134.7, 133.3, 131.1, 129.6, 129.5, 129.2, 127.4, 124.3, 118.3, 115.3, 17.7. LC/MS calcd. for C23H17ClN3O, 386 [M +H]+; found 386.
3-Chloro-5-cyclohexylquinolino[3, 4-b]quinoxalin-6(5H)-one (20e): Yellow solid, yield 37%. 1H NMR (400 MHz, CDCl3): δ 8.91 (d, 1H, J=8.5 Hz), 8.38 (d, 1H, J=8.4 Hz), 8.19 (d, 1H, J=8.5 Hz), 7.90 (s, 1H), 7.82 (s, 1H), 7.57 (s, 1H), 7.34 (dd, 1H, J=8.5, 1.5 Hz), 2.79 (d, 2H, J=11.0 Hz), 2.02-1.78 (m, 6H), 1.50 (dd, 3H, J=21.9, 11.5 Hz). 13C NMR (100 MHz, CDCl3): δ 160.6, 144.3, 144.2, 143.1, 140.1, 138.1, 137.6, 132.7, 130.9, 130.7, 129.1, 127.6, 123.3, 118.9, 115.5, 59.2, 28.8, 26.6, 25.4. LC/MS calcd. for C21H19ClN3O, 364 [M+H]+; found 364.
5-Cyclohexyl-9, 10-dimethylquinolino[3, 4-b]quinoxalin-6(5H)-one (20f): Yellow solid, yield 46%. 1H NMR (400 MHz, CDCl3): δ 9.00 (d, 1H, J=7.9 Hz), 8.15 (s, 1H), 7.99 (s, 1H), 7.63 (d, 2H, J=4.1 Hz), 7.47-7.31 (m, 1H), 2.82 (d, 2H, J=11.0 Hz), 2.56 (d, 6H, J=6.8 Hz), 2.01-1.69 (m, 7H), 1.56-1.44 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.0, 144.2, 144.1, 143.6, 142.2, 141.8, 139.0, 131.5, 129.4, 127.9, 126.1, 122.9, 120.7, 115.3, 58.7, 28.9, 26.7, 25.5, 20.8, 20.6. LC/MS calcd. for C23H24N3O [M+H]+, 358; found 358.
AcknowledgementsThe authors would like to thank the Chongqing Research Program of Basic Research and Frontier Technology (Nos. cstc2015jcyjA1328 and cstc2015zdcy-ztzx0191), the Scientific Research Foundation of Chongqing University of Arts and Sciences (Nos. R2013XY01 and R2013XY02), Sichuan Provincial Science Fund for Distinguished Young Scholars (No. 2015JQO055). We would also like to thank Ms. H.Z. Liu for obtaining the LC/MS and NMR data.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.10.027.
| [1] |
(a) E. Ruijter, R. Scheffelaar, R.V. Orru, Multicomponent reaction design in the quest for molecular complexity and diversity, Angew. Chem. Int. Ed. 50(2011) 6234-6246; (b) R.K. Li, Q.L. Yang, Y. Liu, et al., A novel and green synthesis of indolone-Namino acid derivatives via the Passerini three-component reactions in water, Chin. Chem. Lett. 27(2016) 345-348. |
| [2] |
(a) C.G. Neochoritis, J. Zhang, A. Dömling, Leuckart-Wallach approach to sugar isocyanides and its iMCRs, Synthesis 47(2015) 2407-2413; (b) Z. Hossaini, Solvent-free synthesis of substituted five membered heterocycles:one-pot reaction of primary amine and alkyl propiolate or isothiocyanate in the presence of oxalyl chloride, Chin. Chem. Lett. 25(2014) 159-162; (c) U.K. Sharma, N. Sharma, D.D. Vachhani, E.V.V. Eycken, Metal-mediated post-Ugi transformations for the construction of diverse heterocyclic scaffolds, Chem. Soc. Rev. 44(2015) 1836-1860; (d) G. Koopmanschap, E. Ruijter, R.V. Orru, Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics, Beilstein J. Org. Chem. 10(2014) 544-598. |
| [3] |
(a) B.H. Rotstein, S. Zaretsky, V. Rai, A.K. Yudin, Small heterocycles in multicomponent reactions, Chem. Rev. 114(2014) 8323-8359; (b) Z. Hossaini, F. Rostami-Charati, S. Seyfi, M. Ghambarian, Multicomponent reactions for the synthesis of functionalized 1, 4-oxathiane-3-thiones under microwave irradiation in water, Chin. Chem. Lett. 24(2013) 376-378; (c) G.R. Kuethe, J.T. Kuethe, Practical methodologies for the synthesis of indoles, Chem. Rev. 106(2006) 2875-2911. |
| [4] |
(a) A. Dömling, I. Ugi, Multicomponent reactions with isocyanides, Angew. Chem. Int. Ed. 39(2000) 3168-3210; (b) A. Dömling, Recent developments in isocyanide based multicomponent reactions in applied chemistry, Chem. Rev. 106(2006) 17-89. |
| [5] | P. Patil, J. Zhang, K. Kurpiewska, J. Kalinowska-Tłuścik, A. Dömling, Hydrazine in the Ugi tetrazole reaction. Synthesis 48 (2016) 1122–1130. DOI:10.1055/s-00000084 |
| [6] | A. Dömling, W. Wang, K. Wang, Chemistry and biology of multicomponent reactions. Chem. Rev. 112 (2012) 3083–3135. DOI:10.1021/cr100233r |
| [7] | N. Sharma, Z. Li, U.K. Sharma, E.V. Van der Eycken, Facile access to functionalized spiro[indoline-3, 2'-pyrrole]-2, 5'-diones via post-Ugi domino Buchwald-Hartwig/Michael reaction. Org. Lett. 16 (2014) 3884–3887. DOI:10.1021/ol5019079 |
| [8] | C. Kalinski, M. Umkehrer, J. Schmidt, A novel one-pot synthesis of highly diverse indole scaffolds by the Ugi/Heck reaction. Tetrahedron Lett. 47 (2006) 4683–4686. DOI:10.1016/j.tetlet.2006.04.127 |
| [9] | Z. Xu, F. De Moliner, A.P. Cappelli, C. Hulme, Aldol reactions in multicomponent reaction based domino pathways:a multipurpose enabling tool in heterocyclic chemistry. Org. Lett. 15 (2013) 2738–2741. DOI:10.1021/ol401068u |
| [10] | Z. Xu, F. De Moliner, A.P. Cappelli, C. Hulme, Ugi/aldol sequence:expeditious entry to several families of densely substituted nitrogen heterocycles. Angew. Chem. Int. Ed. 51 (2012) 8037–8040. DOI:10.1002/anie.v51.32 |
| [11] | A. Kumar, Z. Li, S.K. Sharma, V.S. Parmar, E.V. Van der Eycken, An expedient route to imidazo[1, 4] diazepin-7-ones via a post-Ugi gold-catalyzed heteroannulation. Org. Lett. 15 (2013) 1874–1877. DOI:10.1021/ol400526a |
| [12] | B.V. Nicholas, F. Bai, C. Perez, Phenazine antibiotic inspired discovery of potent bromophenazine antibacterial agents against Staphylococcus aureus and Staphylococcus epidermidis. Org. Biomol. Chem. 12 (2014) 881–886. DOI:10.1039/c3ob42416b |
| [13] | M. Conda-Sheridan, L. Marler, E.J. Park, Potential chemopreventive agents based on the structure of the lead compound 2-bromo-1-hydroxyphenazine, isolated from streptomyces species, strain CNS284. J. Med. Chem. 53 (2010) 8688–8699. DOI:10.1021/jm1011066 |
| [14] | W. Zeghida, J. Debray, S. Chierici, P. Dumy, M. Demeunynck, Concise synthesis of 2-amino-4(3H)-quinazolinones from simple (hetero) aromatic amines. J. Org. Chem. 73 (2008) 2473–2475. DOI:10.1021/jo7026883 |
| [15] | A.C. Shekhar, P.S. Rao, B. Narsaiah, D.A. Allanki, P.S. Sijwali, Emergence of pyrido quinoxalines as new family of antimalarial agents. Eur. J. Med. Chem. 77 (2014) 280–287. DOI:10.1016/j.ejmech.2014.03.010 |
| [16] | W. Pendergast, J.V. Johnson, S.H. Dickerson, Benzoquinazoline inhibitors of thymidylate synthase:enzyme inhibitory activity and cytotoxicity of some 3-amino-and 3-methylbenzo[f]quinazolin-1(2H)-ones. J. Med. Chem. 36 (1993) 2279–2291. DOI:10.1021/jm00068a004 |
| [17] | J.W. Chern, P.L. Tao, K.C. Wang, Studies on quinazolines and 1, 2, 4-benzothiadiazine 1. 1-dioxides. 8.1, 2 Synthesis and pharmacological evaluation of tricyclic fused quinazolines and 1, 2, 4-benzothiadiazine 1, 1-dioxides as potential alpha1-adrenoceptor antagonists. J. Med. Chem. 41 (1998) 3128–3141. DOI:10.1021/jm970159v |
| [18] | J.A. Grosso, E.D. Nichols, J.D. Kohli, D. Glock, Synthesis of 2-(alkylamino)-5, 6-and -6, 7-dihydroxy-3, 4-dihydroquinazolines and evaluation as potential dopamine agonists. J. Med. Chem. 25 (1982) 703–708. DOI:10.1021/jm00348a018 |
| [19] | G. Song, S. Li, Z. Yang, Microwave-assisted synthesis of fused piperazinebenzimidazoles via a facile, one-pot procedure. Tetrahedron Lett. 56 (2015) 4616–4618. DOI:10.1016/j.tetlet.2015.06.035 |
| [20] | T. Ou, Y. Lu, J. Tan, G-quadruplexes:targets in anticancer drug design. ChemMedChem 3 (2008) 690–713. DOI:10.1002/(ISSN)1860-7187 |
| [21] | L. El Kaïm, L. Grimaud, Isocyanide Chemistry:Ugi and Passerini Reactions with Carboxylic Acid Surrogates, Wiley-VCH, Weinheim, 2012, pp. 159-194. |
| [22] | M. Avaz, G. Martinez-Ariza, C. Hulme, A robust protocol for the synthesis of quinoxalines and 5H-benzo[e][1, 4] di-azepines via the acidless Ugi reaction. Synlett 25 (2014) 1680–1684. DOI:10.1055/s-00000083 |
| [23] | S. Pan, B. List, Catalytic three-component Ugi reaction. Angew. Chem. Int. Ed. 47 (2008) 3622–3625. DOI:10.1002/(ISSN)1521-3773 |
| [24] | Z. Chen, J. Zhang, D. Tang, Z. Xu, Synthesis of fused benzimidazole-quinoxalinones via UDC strategy and following the intermolecular nucleophilic substitution reaction. Tetrahedron Lett. 55 (2014) 2742–2744. DOI:10.1016/j.tetlet.2014.03.063 |
| [25] | Z. Xu, F. De Moliner, A.P. Cappelli, C. Hulme, Aldol reactions in multicomponent reaction based domino pathways:a multipurpose enabling tool in heterocyclic chemistry. Org. Lett. 15 (2013) 2738–2741. DOI:10.1021/ol401068u |
| [26] | D. Bhattacharya, S. Mitra, P. Chattopadhyay, A rapid one pot synthetic approach towards imidazole fused benzodiazepinone using Ugi reaction strategy. Synthesis 47 (2015) 2294–2298. DOI:10.1055/s-00000084 |