 2017, Vol. 28
2017, Vol. 28 



b Institute of Pharmacy and Pharmacology, Learning Key Laboratory for Pharmacoproteomics University of South China, Hengyang 421001, China
In the past decade, transition-metal-catalyzed C—H bond functionalization reactions have attracted great attention and significant achievements have been made on these topics. Among them, the application of directing groups show great regioselectivity and become the most prevalent strategy for efficient C—H activation [1-9]. Ester is a kind of very important and common functional groups in natural products, drug molecules and practical materials. Therefore the development of esters as directing group would be very important and practical. However, there are only very few examples in literature compared to the dominant directing groups like amides, carboxylic acid, pyridine etc., which is probably due to the weak coordinating feature of ester: for instance, Chang and co-workers reported an ester as an efficient directing group in the Rh (Ⅲ)-catalyzed olefination of aromatic ester [10], and later on the same group developed an Ir (Ⅲ)-catalyzed direct amidation of aromatic esters [11]. In a related contribution, Graczyk et al. disclosed a Ru (Ⅱ)-catalyzed oxidative alkenylations with aromatic esters [12]. In addition, Shan et al. described a Pd (Ⅱ)-catalyzed regioselective C—H oxygenation of benzoates [13]. Recently, Li et al. developed an ortho-olefination of phenylacetic Weinreb amides, esters and ketones [14]. Furthermore, a Pd (Ⅱ)-catalyzed ortho-olefination of phenyl acetic and propionic acid esters was described by Hu et al. [15]. The ortho-olefination product of phenylacetic esters is a key intermediate forthe syntheses of 2-tetralone derivatives, which are usually needed multi-step synthesis in existing methods [16]. Herein, we report a Pd (Ⅱ)-catalyzed ortho-olefination of aromatic acetic esters as complementary protocol to precedent work, which characterizes with a broader substrate scope and less reaction time.
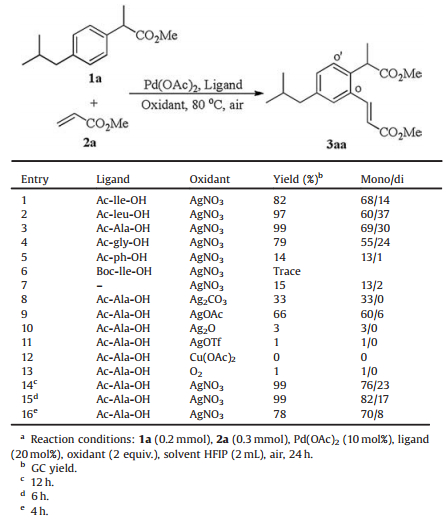
2. Results and discussionIbuprofen methyl ester 1a and methyl acrylate 2a were chosen as the substrates to test the feasibility (Table 1). Starting with Pd (OAc)2 as the catalyst, Ac-lle-OH as the ligand and AgNO3 as oxidant in HFIP (1, 1, 1, 3, 3, 3-hexafluoro-2-propanol), to our delight, the desired product 3aa was formed in 82% GC yield that displayed general mono-selectivity (mono: di=68:14) (Table 1, entry 1). Further ligand screening suggested that Ac-Ala-OH was the optimal one over Ac-lle-OH, Ac-leu-OH, Ac-gly-OH, Ac-ph-OH and Boc-lle-OH (Table 1, entries 1-6). Control experiment without ligand was performed accordingly, and much lower yield was obtained with poor selectivity, which revealed the importance of ligand for the transformation (entry 7). Subsequently, oxidant optimization demonstrated that AgNO3 was the best one among Ag2CO3, AgOAc, Ag2O, AgOTf, Cu (OAc)2 and O2 (Table 1, entries 8-13). It is noteworthy that significant improvement of monoselectivity was observed when the reaction time was decreased from 24 h to 4 h (mono:di up to 70:8), however, the substrate did not completely consumed at 4 h (Table 1, entries 14-16). So we chose 6 h as the optimized reaction time with high yield and better monoselectivity. It is of note that other solvent system, such as DMSO, DMF, t-amyl-OH and 2, 2, 2-trifluoroethanol could not facilitate this reaction. It needs to be pointed out that compared with Yu's method [14], our method has obvious advantages, much short reaction time (6h vs. 48 h), better mono/di ratio, mild reaction temperature etc.
|
|
Table 1 Development of optimized conditions for ortho-olefination.a |
With the optimized conditions in hand, we next turned our attention to the substrate scope of this palladium-catalyzed ortho-olefination. Avery broad range of substituted aromatic acetic esters (1a-1m, Scheme 1) with 2a could be smoothly converted into the corresponding ortho-olefination products with high regioselectivity. Firstly, esters of ibuprofen were demonstrated to be tolerated well under our standard conditions, and methyl, ethyl, propyl and pentyl esters all provided corresponding products in good yields (3aa-3ca, Scheme 1). Then, naproxen methyl ester (1e, Scheme 1) also was a good substrate to participate in ortho-olefination with 58% yield. Next, a-position of phenylacetic esters has phenyl (1f, Scheme 1) ortwomethyls (1g, Scheme 1) could proceed smoothly to afford ortho-olefination product with high regioselectivity. Both electron-rich and electron-deficient substituents on the aromatic rings were found to be good candidates for this transformation as well (3ha-3ma, Scheme 1), albeit the latter ones giving lower yields (3la-3ma, Scheme 1).

|
Download:
|
| Scheme 1. Substrate scope of aromatic acetic esters. Reaction conditions: 1a (0.2 mmol), 2a (0.3mmol), Pd (OAc)2 (10 mol%), Ac-Leu-OH (20 mol%), AgNO3 (2 equiv.), HFIP (2 mL), air, isolated yield. | |
{kind=link}
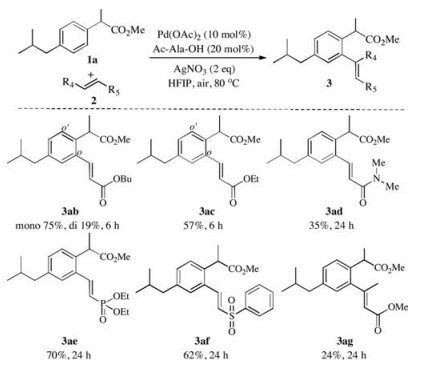
Subsequently, the scopes of alkene reactants were investigated with aromatic acetic ester under the standard conditions (Scheme 2). To our delight, butyl acrylate and ethyl acrylate were reacted to afford the corresponding products in good yields (3ab and 3ac, Scheme 2). ortho-Olefination of aromatic acetic ester 1a with other various olefin coupling partners such as α, β-unsaturated amide, phosphonate, and sulphone led to the corresponding desired products in moderate to good yields (3ad-3af, Scheme 2). Moreover, interestingly, disubstituted olefin 2g was found to be a good candidate for this transformation as well, rendering 3ag 24% yield.

|
Download:
|
| Scheme 2. Substrate scope of alkene. Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), Pd (OAc)2 (10 mol%), Ac-Ala-OH (20 mol%), AgNO3 (2 equiv.), HFIP (2 mL), air, isolated yield. | |
{kind=link}
To our delight, this reaction could be smoothly conducted on gram scales without loss of efficiency (Scheme 3). When ibuprofen methyl ester 1a (5 mmol) or methyl 2-(4-(tert-butyl) phenyl)-acetate 1i (5 mmol) was exposed to the standard condition, 78% of 3aa and 79% of 3ia were obtained, which demonstrated its potential industrial value.

|
Download:
|
| Scheme 3. Scale-up experiments. | |
{kind=link}
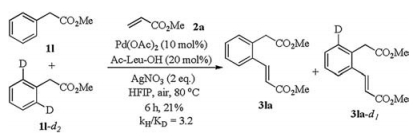
Furthermore, a significant KIE value was observed from isotope effect study (kH/kD=3.2), implying that the mechanism involves Pd-mediated C—H bond cleavage step rather than Friedel-Crafts reaction (Scheme 4).

|
Download:
|
| Scheme 4. Isotope effect study. | |
{kind=link}
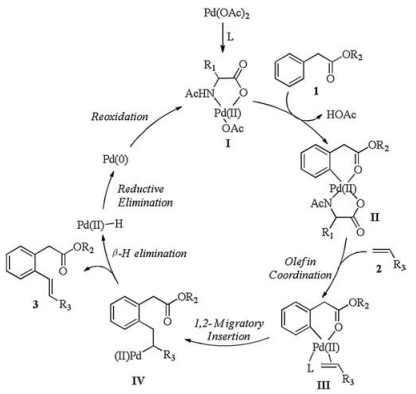
According to the isotope effect study result as well as previous literature reports [17, 18], we gave a proposed mechanism (Scheme 5): aromatic acetic ester 1 directed C—H bond cleavage in presence of catalyst Ⅰ from ligand exchange of Pd (OAc)2. Intermediate Ⅳ was generated from olefin coordination and 1, 2-migratory insertion between intermediate Ⅱ and olefin 2. β-H elimination of intermediate Ⅳ afforded the product 3 and Pd (Ⅱ)-H. The latter underwent reductive elimination to produce Pd (0), then it was oxidized to Pd (Ⅱ) by Ag (Ⅰ) to complete the catalytic cycle (Scheme 5).

|
Download:
|
| Scheme 5. Plausible catalytic cycle for ortho-olefination of aromatic acetyl ester. | |
{kind=link}
3. Conclusion
In conclusion, a Pd (Ⅱ)-catalyzed ortho-olefination of aromatic acetic esters with a wide range of olefin coupling partners has been disclosed. This method features with excellent functional group tolerance, good yields, mild reaction conditions, good scalability as well as high chemo-and regioselectivity. And this work is complementary to the existing methods to synthesize more significant molecule.
4. ExperimentalAll experiments were conducted with a schlenk tube. Flash column chromatography was performed over silica gel (200-300 mesh). 1H NMR and 13C NMR spectra were recorded at ambient temperature using Bruker AVIII-500M spectrometers, chemical shifts (in ppm) were referenced to CDCl3 (δ 7.26) and DMSO-d6(δ 2.50) as internal standards. 13C NMR spectra were obtained by using the same NMR spectrometers and were calibrated with CDCl3 (δ 77.0), DMSO-d6 (δ 39.6). Unless otherwise noted, materials obtained from commercial suppliers were used without further purification.
General procedure for the ortho-olefination of aromatic acetyl ester: In air, a 25 mL sealed tube was charged with methyl 2-(4-isobutylphenyl) propanoate (44 mg, 0.2 mmol, 1 equiv.), methyl acrylate (25.8 mg 0.3 mmol, 1.5 equiv.), Pd (OAc)2 (4.5 mg, 10 mol%), Ac-Ala-OH (5.3 mg, 20 mol%), AgNO3 (68 mg, 0.4 mmol, 2 equiv.) and HFIP (2 mL). The resulting solution was stirred at 80 ℃ under air for 6 h. Upon completion of the reaction, the mixture was cooled down to room temperature and the reaction mixture was diluted with ethyl acetate, filtered through a silica gel plug, rinsed with ethyl acetate, and concentrated in vacuo. The crude reaction mixture was purified on silica gel (petroleum ether:ethyl acetate=30:1-10:1) to afford the desired products 3aa-mono (82%) and 3aa-di (17%).
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (No. 21202049), the Recruitment Program of Global Experts (1000 Talents Plan), the Natural Science Foundation of Fujian Province (No. 2016J01064), Fujian Hundred Talents Plan and Program of Innovative Research Team of Huaqiao University (No. Z14X0047) are gratefully acknowledged. We also thank Instrumental Analysis Center of Huaqiao University for analysis support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.11.023.
| [1] | L. Ackermann, R. Vicente, A.R. Kapdi, Transition-metal-catalyzed direct arylation of (hetero) arenes by C-H bond cleavage. Angew. Chem. Int. Ed. 48 (2009) 9792–9826. DOI:10.1002/anie.v48:52 |
| [2] | X. Chen, K.M. Engle, D.H. Wang, J.Q. Yu, Palladium (Ⅱ)-catalyzed C-H activation/C-C cross-coupling reactions:versatility and practicality. Angew. Chem. Int. Ed. 48 (2009) 5094–5115. DOI:10.1002/anie.v48:28 |
| [3] | T.W. Lyons, M.S. Sanford, Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 110 (2010) 1147–1169. DOI:10.1021/cr900184e |
| [4] | W. Shi, C. Liu, A. Lei, Transition-metal catalyzed oxidative cross-coupling reactions to form C-C bonds involving organometallic reagents as nucleophiles. Chem. Soc. Rev. 40 (2011) 2761–2776. DOI:10.1039/c0cs00125b |
| [5] | C. Liu, H. Zhang, W. Shi, A. Lei, Bond formations between two nucleophiles:transition metal catalyzed oxidative cross-coupling reactions. Chem. Rev. 111 (2011) 1780–1824. DOI:10.1021/cr100379j |
| [6] | K.M. Engle, T.S. Mei, M. Wasa, J.Q. Yu, Weak coordination as a powerful means for developing broadly useful C-H functionalization reactions. Acc. Chem. Res. 45 (2012) 788–802. DOI:10.1021/ar200185g |
| [7] | J. Ding, Y. Guo, L.Y. Shao, Palladium-catalyzed multi-acetoxylation of 1, 3-disubstituted 1H-pyrazole-5-carboxylates via direct C (sp2)-H or C (sp3)-H bond activation. Chin. Chem. Lett. 27 (2016) 1617–1621. DOI:10.1016/j.cclet.2016.04.007 |
| [8] | X. Lu, B. Xiao, R. Shang, L. Liu, Synthesis of unnatural amino acids through palladium-catalyzed C (sp3)-H functionalization. Chin. Chem. Lett. 27 (2016) 305–311. DOI:10.1016/j.cclet.2015.12.021 |
| [9] | G. Shan, G.Y. Huang, Y. Rao, H. Zhang, Palladium-catalyzed ortho-selective C-H bond chlorination of aromatic ketones. Chin. Chem. Lett. 26 (2015) 1236–1240. DOI:10.1016/j.cclet.2015.07.011 |
| [10] | S.H. Park, J.Y. Kim, S. Chang, Rhodium-catalyzed selective olefination of arene esters via C-H bond activation. Org. Lett. 13 (2011) 2372–2375. DOI:10.1021/ol200600p |
| [11] | J. Kim, S. Chang, Iridium-catalyzed direct C-H amidation with weakly coordinating carbonyl directing groups under mild conditions. Angew. Chem. Int. Ed. 53 (2014) 2203–2207. DOI:10.1002/anie.201310544 |
| [12] | K. Graczyk, W. Ma, L. Ackermann, Oxidative alkenylation of aromatic esters by ruthenium-catalyzed twofold C-H bond cleavages. Org. Lett. 14 (2012) 4110–4113. DOI:10.1021/ol301759v |
| [13] | G. Shan, X. Yang, L. Ma, Y. Rao, Pd-catalyzed C-H oxygenation with TFA/TFAA:expedient access to oxygen-containing heterocycles and late-stage drug modification. Angew. Chem. Int. Ed 51 (2012) 13070–13074. DOI:10.1002/anie.201207458 |
| [14] | G. Li, L. Wan, G. Zhang, Pd (Ⅱ)-catalyzed C-H functionalizations directed by distal weakly coordinating functional groups. J. Am. Chem. Soc. 137 (2015) 4391–4397. DOI:10.1021/ja5126897 |
| [15] | J. Hu, M. Guan, J. Han, Palladium-catalyzed ortho-olefination of phenyl acetic and phenyl propylacetic esters via C-H bond activation. J. Org. Chem. 80 (2015) 7896–7904. DOI:10.1021/acs.joc.5b00775 |
| [16] | Á. Gorka, B. Czuczai, P. Szoleczky, Convenient synthesis of 7, 8-dimethoxytetralin-2-one. Synth. Commun. 35 (2005) 2371–2378. DOI:10.1080/00397910500187753 |
| [17] | K.M. Engle, D.H. Wang, J.Q. Yu, Ligand-accelerated C-H activation reactions:evidence for a switch of mechanism. J. Am. Chem. Soc. 132 (2010) 14137–14151. DOI:10.1021/ja105044s |
| [18] | K.M. Engle, P.S. Thuy-Boun, M. Dang, J.Q. Yu, Ligand-accelerated cross-coupling of C (sp2) H bonds with arylboron reagents. J. Am. Chem. Soc. 133 (2011) 18183–18193. DOI:10.1021/ja203978r |