 2017, Vol. 28
2017, Vol. 28 



b Key Laboratory of Drug Targeting and Drug Delivery Systems of Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu 610041, China
Neuromuscular blocking agents (NMBAs), also known as skeletal muscle relaxants (MRs), can induce paralysis by preventing neurotransmitter acetylcholine (ACh) from binding to motor endplates on skeletal muscles, and thus relax it. NMBAs, one of the three major classes of drugs routinely used in modern anaesthesia [1], are widely used in surgery nowadays. According to the mechanisms of blocking action, NMBAs are divided into depolarizing and non-depolarizing agents. Among the depolarizing agents which interrupts the normal respondence of the nicotinic ACh receptor (nAChRs) by depolarizes motor end plates, suxamethonium is the only drug still being used in clinical practice. However, the utility of suxamethonium is limited for the mechanism-caused side effects, such as potassium release and muscle fasciculation pain [2]. The non-depolarizing NMBAs, also called competitive antagonists, can compete with ACh for nAChR without producing depolarization or triggering any intrinsic activity. Especially the steroidal non-depolarizing NMBAs can mimic the ACh binding to receptor with ACh-like moiety built on the androstane core, and they are among the most important clinically used MRs [3].
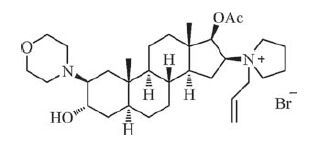
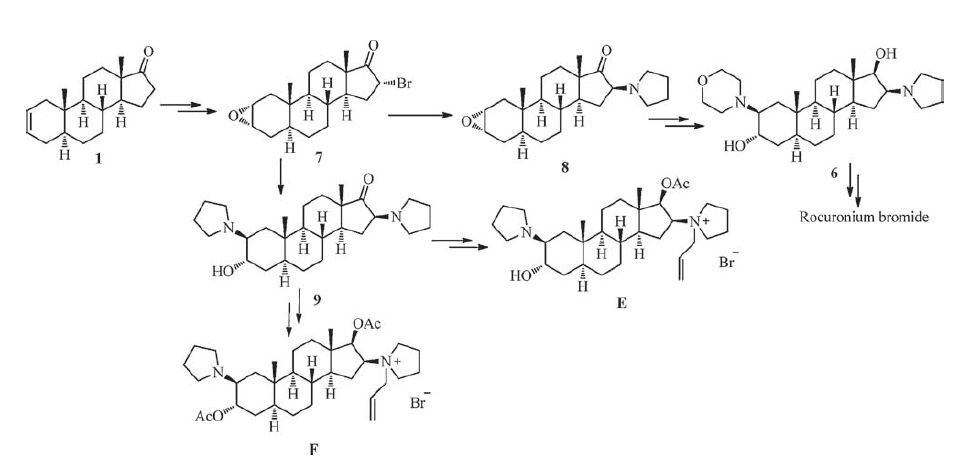
Rocuronium bromide (Fig. 1), a typical non-depolarizing steroidal muscle relaxant developed and produced by the Organon (US), achieves rapid onset and short duration of action, little accumulation effect or side effect in vivo, and it is currently the most widely used muscle relaxant [4-6]. Due to the excellent clinical effect and broad application, rocuronium bromide has been widely concerned in the chemical synthesis field. Generally, the traditional routes for synthesis of rocuronium bromide are mostly performed through a ring-opening of 2α, 3α-epoxy steroids with morpholine after introducing 16β-pyrrolidinyl in low yields [7-15]. It is noteworthy that the traditional methods provide eightimpurities described in the European Pharmacopoeia 8.0 (EP 8.0) which cannot be avoided. Additionally, we have imitated the methods of literature and found the impurity E and F can hardly removefrom the target compound in the process. Imaginably, when introducing 16β-pyrrolidinyl by using pyrrolidine to substitute 16-bromo, it may trigger the ring-opening of 2α, 3α-epoxy and eventually lead to the generation of impurity E and F (Fig. 2).

|
Download:
|
| Figure 1. The structure of rocuronium bromide | |

|
Download:
|
| Figure 2. Traditional synthetic route for rocuronium bromide. | |
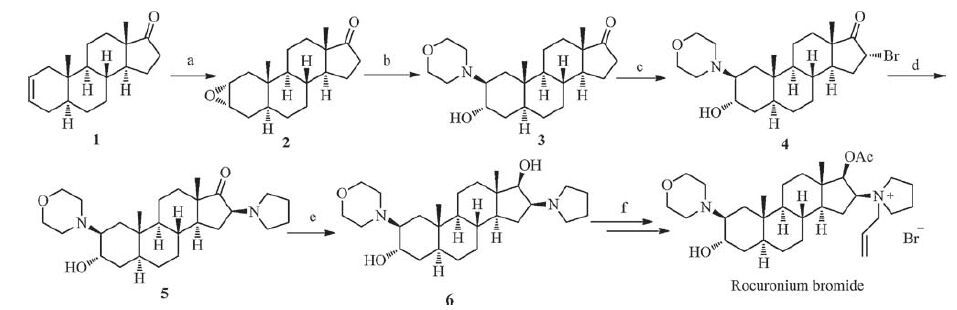
Due to our interests on the steroid structures [16-19], we are committed to find an alternative synthetic route affording rocuronium bromide with the improved overall yield and avoiding the generation of the impurities. Herein, we introduce a new synthetic route for 2β- (4-morpholinyl) -16β- (1-pyrrolidinyl) -5aandrostan-3α, 17β-diol (6), the key intermediate which can easily provide rocuronium bromide systematically, in high yield through a ring-opening of epoxide followed by introducing pyrrolidine to substitute 16a-bromo group, and this method could significantly avoid the generation of the impurities E and F.
2. Results and discussionThe synthetic protocol of the rocuronium bromide is depicted in Scheme 1. Firstly, 5α-androstan-2-en-17-one (1) was chosen as the starting material and the double bond was epoxidized using HCOOH/H2O2 to give epoxide 2 with a good yield (up to 88.5%) [20]. Other conditions for epoxidation [21-24], such as m-CPBA and CH3COOOH/CH3COONa·3H2O, showed less effective. Then, the conditions for ring-opening of 2α, 3α-epoxy-5α-androstan-17-one (2) with morpholine were screened. Based on the previous reports [25, 26], we employed TsOH and ZnCl2 to promote this transformation. However, the results were not as satisfying as expecting (Table 1, entries 2, 3). Thus, we tried to optimize the reaction by screening different types of Lewis acids such as ZnI2, CuBr2, AlCl3, FeCl3, Pd (OAc) 2, etc. Among these catalysts, FeCl3 exhibited the best activity (Table 1, entries 3-14). Notably, the use of adequate H2O was necessary, and the reaction yield decreased when the amount of H2O was decreased (Table 1, entries 9, 15-17). In addition, decreasing the amount of morpholine to 20 equiv. proved to be more effective (Table 1, entries 9, 18-22). Furthermore, we found that the yield of compound 3 was not decreased when reaction time was shortened to 24 h (Table 1, entries 22-24). Unsurprisingly, when the reaction temperature was reduced to 100 ℃ and even lower to 80 ℃ or 60 ℃, the yield of the desired product 3 dropped to 78%, 72% and 46%, respectively (Table 1, entries 23, 25-27).

|
Download:
|
| Scheme 1. Synthesis of rocuronium bromide. Reagents and conditions: (a) 30% H2O2, HCOOH, CH2Cl2, r.t., 5 h; (b) morpholine, FeCl3, H2O, reflux, 24 h; (c) CuBr2, CH3OH, reflux, 30 h; (d) pyrrolidine, CH3CN, reflux, 8 h; (e) NaBH4, CH3OH,-3℃, 2 h; (f) (1) AcO2, TEA, CH2Cl2, 25 ℃, 22 h; (2) allyl bromide, Na2CO3, CH2Cl2, 40 ℃, 22 h. | |
|
|
Table 1 Screening of conditions of the ring-opening of compound 2.a |

{kind=link}
{kind=link}
{kind=link}
Subsequently, our research focused on the conditions of the a-bromination of compound 3. Our initial experiment was carried out with compound 3 and 2 equiv. of cupric bromide complex in methanol at reflux for 8 h as literature described [27, 28], but only affording the desired product compound 4 in 37% yield together with low conversion (Table 2, entry 1). Fortunately, the complete transformation of the material compound 3 was fulfilled by simply prolonging the reaction time and increasing the amount of cupric bromide. The reaction performed well in the presence of 4 equivalent of cupric bromide for 30h and product 4 could be obtained in 93% yield (Table 2, entries 2-8). It should be noted that the use of 4 equiv. of cupric bromide is necessary to achieve a satisfactory yield, but further increases in the amount of cupric bromide would not improve the yield. Similarly, 30h of the reaction time can be the most effective and appropriate. As decreasing the reaction temperature resulted in dramatically decreasing yields (Table 2, entries 6, 9, 10), we assumed whether solvents with higher boiling point might increase the yields or decrease the reaction time. However, experiments showed the opposite results (Table 2, entries 11-14). Different bromide reagents were also tried [29-34], and cupric bromide was still the most suitable one (Table 2, entries 15-20).
|
|
Table 2 Screening of conditions of the α-bromination of compound 3.a |
Furthermore, compound 4 was subjected to substitution of 16α-bromogroup by pyrrolidine (5 equiv. in MeCNat reflux for 8h) to provided 16β- (1-pyrrolidinyl) -steroid (5) in 83% yield. The attack of pyrrolidine on 16-carbon with the steric effects leads to inversion of configuration. It is interesting that whether to extend or reduce the reaction time would result in remarkable decrease in the yield. Finally reduction of 17-carbonyl to 17-β-hydroxyl was achieved by NaBH4 (3.1 equiv.) in methanol at -3 ℃ to afford the key intermediate 6 with 92% yield, which could easily provide rocuronium bromide systematically as literature described [12, 13]. In the synthesis process of the known compound 6 from 5, the bond between the chiral centers and other atom (N, O or H) could not be broken, so the configurations of chiral centers were not changed (besides the C17-OH of 6). In consideration that the compound 6 had a consistent date (NMR and optical rotation) with the reported literatures, hence, the absolute stereochemistry of 5 was concluded.
3. ConclusionHence, we have demonstrated a facile and efficient strategy to design and synthesize rocuronium bromide, starting from 5α-androstan-2-en-17-one (1). In this novel synthetic route, 2β- (4-morpholinyl) -16β- (1-pyrrolidinyl) -5α-androstan-3α, 17β-diol (6), as key intermediate, can be successfully obtained by a ringopening of epoxide followed by introducing pyrrolidine with 57.8% overall yield, while previously reported yield was in the range of 19.8-55.3% [7-15]. Moreover, this new approach would not produce the impurities E and F that are difficult to remove by traditional ways. Thus, this protocol offers advantages of operational simplicity, pleasing yield and purity of product.
4. Experimental 4.1. GeneralAll the reactions were monitored by thin-layer chromatography (TLC) and were visualized using iodine vapor. Most chemicals and solvents were analytical grade and used without further purification. Thin layer chromatography (TLC) was performed with precoated silica gel GF254 (0.2 mm), while column chromatography was performed using silica gel (100-200 mesh). The melting point was measured on an YRT-3 melting point apparatus (Shantou Keyi instrument & Equipment Co., Ltd., Shantou, China). The optical rotation was measured on an Anton Paar MCP 200 polarimeter (λ= 589 nm). NMR spectra were taken on a Varian INOVA 400 or 600 MHz (Varian, Palo Alto, CA, USA) using CDCl3 as solvent. Chemical shifts were expressed in δ (ppm), with tetramethylsilane (TMS) functioning as the internal reference, and coupling constants (J) were expressed in Hz. High resolution mass spectroscopy data of the product were collected on a Waters Micromass GCT or a Bruker Apex IV FTMS instrument.
4.2. Synthesis of 2α, 3α-epoxy-5α-androstan-17-one (2)In a 250 mL round-bottom flask, compound 1 (15.0 g, 55 mmol) was dissolved in CH2Cl2 (70 mL), then 30% H2O2 (18 mL, 179 mmol) was added dropwise to the stirred reaction followed by the addition of formic acid (6 mL, 159 mmol). The stirring was continued at room temperature for 5 h while the solution became colorless from starting light yellow. Under ice bath, 1 mol/L NaOH (aq) was added to adjust the mixture to pH 12, and then separate the organic layer and washed with water (3 × 50 mL). After drying over Na2SO4 and filtering, the solvent was removed under reduced pressure and the residue was purified by recrystallization with methanol to give product 2 (14.06 g, 88.5%) as white solid. Mp: 120-124 ℃ (Lit. [35] 123-126 ℃); [α]D20+ 108 (c 1, CHCl3) (Lit. [35] +104, CHCl3). 1H NMR (400 MHz, CDCl3) : δ 0.78 (s, 3H, 18-CH3), 0.84 (s, 3H, 19-CH3), 2.01-2.11 (m, 1H, 16α-H), 2.39-2.46 (m, 1H, 16β-H), 3.10-3.13 (m, 1H, 2β-H), 3.16 (br, 1H, 3β-H). HRMS: (ESI+) calcld. for C19H28O2Na+ [M+Na]+ 311.1982, found 311.1985.
4.3. Synthesis of 3α-hydroxy-2β- (4-morpholinyl) -5α-androstan-17- one (3)Compound 2 (2.00 g, 6.93 mmol) and FeCl3 (2.25 g, 13.87 mmol) were added into deionized water (25 mL) and the solution was stirred for 30 min at room temperature. Under ice bath, morpholine (12 mL, 137 mmol) was added dropwise over 30 min, and the mixture was heated at reflux for 24 h. The reaction was cooled and filtered, and the filter cake was washed with EtOAc (3 × 20 mL). The combined organic layer was washed with water till morpholine was removed completely. After drying over Na2SO4, the solvent was removed under reduced pressure and the residue was purified by recrystallization with acetone to give compound 3 (2.36 g, 90.8%) as white solid. Mp: 168-172 ℃ (Lit. [25] 171-173 ℃); [α]D20+ 138 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) : δ 0.86 (s, 3H, 18- CH3), 0.89 (s, 3H, 19-CH3), 2.02-2.11 (m, 1H, 16α-H), 2.41-2.65 (m, 6H, 16β-H and 2α-H and 2-NCH2 × 2), 3.36 (s, 1H, OH), 3.66-3.74 (m, 4H, 2-OCH2 × 2), 3.85-3.90 (m, 1H, 3b-H). HRMS: (ESI+) calcld. for C23H37NO3Na+ [M+Na]+ 398.2666, found 398.2670.
4.4. Synthesis of 3α-hydroxy-2β- (4-morpholinyl) -16α-bromo-5α-androstan-17-one (4)To a solution of compound 3 (550 mg, 1.46 mmol) in anhydrous methanol (20 mL) at a dried 50 mL round-bottom flask was added CuBr2 (1.31 g, 5.84 mmol) in portions. The mixture was heated to reflux for 30 h under argon. Then the mixture was cooled to room temperature and concentrated in vacuum. The residue was dissolved in 12.5% ammonia solution (40 mL) and stirred for 10 min, and then extracted with EtOAc (30 mL). The organic layer was separated and washed with another 12.5% ammonia solution (40 mL), water (3 × 40 mL) and brine (30 mL), after that dried over Na2SO4. The solvent was evaporated in vacuum and the residue was purified by recrystallization with petroleum ether and acetone to afford compound 4 (616 mg, 92.8%) as white solid. Mp: 141- 145 ℃; [α]D20+ 101 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) : δ 0.90 (s, 3H, 18-CH3), 1.07 (s, 3H, 19-CH3), 2.11-2.24 (m, 1H, 15α-H), 2.52- 2.69 (m, 6H, 15β-H and 2α-H and 2-NCH2 × 2), 3.39 (br, 1H, OH), 3.74 (s, 4H, 2-OCH2 × 2), 3.90-3.93 (m, 1H, 3b-H), 4.09 (t, 1H, 16aH, J = 8.8 Hz). 13C NMR (150 MHz, CDCl3) : δ 14.20, 16.68, 20.44, 27.77, 29.65, 30.27, 32.37, 32.55, 33.99, 34.20, 34.29, 35.91, 38.43, 46.29, 47.79, 47.85, 55.72 (C × 2), 63.64, 64.92, 67.24 (C × 2), 213.30. HRMS: (ESI+) calcld. for C23H36BrNO3Na+ [M+Na]+ 476.1771, found 476.1768.
4.5. Synthesis of 3α-hydroxy-2β- (4-morpholinyl) -16β- (1- pyrrolidinyl) -5α-androstan-17-one (5)Under argon protection, pyrrolidine (185μL, 2.2 mmol) was taken into to a dried 25 mL two-neck round bottom flask in the stirred solution of compound 4 (200 mg, 0.44 mmol) in anhydrous acetonitrile (10 mL). The entire mixture was heated under reflux for 8 h. Then the reaction was cooled to room temperature and concentrated in vacuo. The residue was added with 2% HCl aqueous solution (15 mL) and extracted with EtOAc (20 mL). The pH value of the aqueous layer was adjusted with saturated sodium carbonate solution to pH 9-10 and then filtered to remove the solid. EtOAc (20 mL) was added into the aqueous layer. After stirring for 10 min, organic layer was separated and washed with deionized water (3 × 20 mL) and brine (20 mL) followed by drying over Na2SO4. The solvent was removed in vacuum and the residue was purified by flash column chromatography to afford compound 5 (164 mg, 83.7%) as light yellow solid. Mp: 177-183 ℃ (Lit. [36] 185 ℃); [α]D20+ 78.6 (c 0.5, CHCl3) (Lit. [36] +79, c 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) : δ 0.86 (s, 3H, 18-CH3), 0.99 (s, 3H, 19-CH3), 2.40- 2.49 (m, 5H, 16-NCH2 × 2, 2α-H), 2.66 (br, 4H, 2-NCH2 × 2), 3.72 (br, 5H, 2-OCH2 × 2 and 16α-H), 3.86-3.92 (m, 1H, 3β-H). HRMS: (ESI+) calcld. for C27H44N2O3Na+ [M+Na]+ 467.3244, found 467.3241.
4.6. Synthesis of 2β- (4-morpholinyl) -16β- (1-pyrrolidinyl) -5α-androstan-3α, 17β-diol (6)To a solution of compound 5 (30.0 mg, 0.0675 mmol) in methanol (2 mL) at a 10 mL round-bottom flask was added NaBH4 (8 mg, 0.21 mmol) carefully at -3 ℃. The mixture was stirred for 2 h at the temperature. CH2Cl2 (10 mL) and deionized water (15 mL) was added, and the mixture was stirred for another 10 min to quench the reaction. The organic layer was separated and washed with water (3×10 mL) and brine (10 mL). After drying over Na2SO4, the solvent was removed to afford compound 6 (27.8 mg, 92.7%) as white solid without further purification. Mp: 215-218 ℃ (Lit. [13] 212-219 ℃); [α]D20+ 86.2 (c 0.5, CHCl3) (Lit. [13] +87.9, c 1.02, CHCl3). 1H NMR (400 MHz, CDCl3) : δ 0.70 (s, 3H, 18-CH3), 0.86 (s, 3H, 19- CH3), 2.42-2.76 (m, 9H, 16-NCH2 × 2, 2α-H and 2-NCH2 × 2), 2.91- 2.95 (m, 1H, 16α-H), 3.38-3.40 (m, 1H, 17α-H), 3.65-3.74 (m, 4H, 2- OCH2 × 2), 3.83-3.89 (m, 1H, 3β-H). HRMS: (ESI+) calcld. for C27H46N2O3Na+ [M+Na]+ 469.3401, found 469.3398.
4.7. Rocuronium bromideAccording to the method of literatures [12, 13], compound 6 was used as material to provide rocuronium bromide as white solid. Mp: 162-166 ℃ (Lit. [13] 161-169 ℃); [α]D20+ 18.9 (c 0.5, CHCl3) (Lit. [13] +18.7, c 1.03, CHCl3). 1H NMR (600 MHz, CDCl3) : δ 0.79 (s, 3H, 18-CH3), 0.85 (s, 3H, 19-CH3), 2.21 (s, 3H, OCH3), 2.56 and 2.68 (br × 2, 5H, 2α-H and 2-NCH2 × 2), 3.73-3.92 (m, 9H, 16-NCH2 × 2, 2-OCH2 × 2 and 3β-H), 4.10-4.19 and 4.29-4.33 (m × 2, 2H, C=C-CH2), 4.53 (br, 1H, OH), 5.21 (d, 1H, J = 8.4 Hz, 17α-H), 5.68-5.72 (m, 2H, C=CH2), 6.12-6.22 (m, 1H, C=CH). 13C NMR (150 MHz, CDCl3) : δ 13.71, 16.45, 20.84, 21.40 (C × 2), 24.52, 27.75, 28.25, 31.30, 33.27, 33.77, 34.57, 35.96, 37.71, 38.43, 43.27, 45.36, 46.90 (C × 2), 49.38, 52.20, 54.99 (C × 2), 61.13, 63.83 (C × 2), 65.36, 66.62, 77.98, 125.89, 128.80, 168.80.
AcknowledgmentWe are grateful for support from the National Natural Science Foundation of China (No. 81373259 & No. 81573286).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.10.026.
| [1] | D.C. Rees, D.R. Hill, Drugs in anesthetic practice. Annu. Rep. Med. Chem. 31 (1996) 41–50. DOI:10.1016/S0065-7743(08)60444-3 |
| [2] | Z. Tuba, S. Maho, E.S. Vizi, Synthesis and structure-activity relationships of neuromuscular blocking agents. Curr. Med. Chem. 9 (2002) 1507–1536. DOI:10.2174/0929867023369466 |
| [3] | H. Hu, P. Liu, X. Hu, 16-Morpholino quaternary ammonium steroidal derivatives as neuromuscular blocking agents:synthesis, biological evaluation and in silico probe of ligand-receptor interaction. Eur. J. Med. Chem. 56 (2012) 332–347. DOI:10.1016/j.ejmech.2012.07.048 |
| [4] | J.T. Pento, J. Castañer, Rocuronium bromide. Drugs Future 19 (1994) 841. DOI:10.1358/dof.1994.019.09.264193 |
| [5] | C. Lee, Conformation, action, and mechanism of action of neuromuscular blocking muscle relaxants. Pharmacol. Ther. 98 (2003) 143–169. DOI:10.1016/S0163-7258(03)00030-5 |
| [6] | D.P. Zlotos, Recent advances in neuromuscular blocking agents. Mini Rev. Med. Chem. 5 (2005) 595–606. DOI:10.2174/1389557054023215 |
| [7] | E.W. Moore, J.M. Hunter, The new neuromuscular blocking agents:do they offer any advantages. Br. J. Anaesth. 87 (2001) 912–925. DOI:10.1093/bja/87.6.912 |
| [8] | F. Chávez, S. Suárez, M.A. Díaz, Sulfuric acid adsorbed on silica gel. A multipurpose acid catalyst. Synth. Commun. 24 (1994) 2325–2339. DOI:10.1080/00397919408019058 |
| [9] | H. Hu, Z. Rao, M. Feng, 3,16-Bisquaternary ammonium steroid derivatives as neuromuscular blocking agents:synthesis and biological evaluation. Steroids 996 (2015) 103–114. |
| [10] | J.A. Mendez, M.A. De La Mora, A. Guillen, et al., Processes for the synthesis of rocuronium bromide, U.S. Patent US20070117975(issued May 24, 2007). |
| [11] | E. Adar, D. Sondack, O. Friedman, et al., Processes for the preparation of rocuronium bromide and intermediates thereof, U.S. Patent US20050159398(issued July 21, 2005). |
| [12] | T. Sleigh, D.S. Savage, I.C. Carlyle, Novel 2β-morpholino-androstane derivatives, U.S. Patent US4894369(issued June 16, 1990). |
| [13] | J.A. Mendez, M.A. De La Mora, A.A. Rodriguez, et al., Process for the synthesis of rocuronium bromide, WIPO Patent WO2007033348A3(issued March 22, 2007). |
| [14] | G. Nadamuni, C.S. Venkatesan, P.M. Senthilkumar, Process for the preparation of rocuronium bromide and intermediate thereof, WIPO Patent WO2009016648A (issued February 5, 2009). |
| [15] | J. Jia, Z. Han, S. Ma, S. Cao, Study on the synthesis and technology of rocuronium bromide. Chem. Res. Appl. 25 (2013) 1417–1421. |
| [16] | Y.L. Wang, L. Hai, L. Guo, Y. Wu, First synthesis of 22-oxachenodeoxycholic acid analogue. Steroids 110 (2016) 70–76. DOI:10.1016/j.steroids.2016.04.004 |
| [17] | Y.G. Yu, Y. He, Y. Zhao, L. Hai, Y. Wu, A simple and convenient synthetic route to Ulipristal acetate. Steroids 78 (2013) 1293–1297. DOI:10.1016/j.steroids.2013.09.009 |
| [18] | X. Cheng, X.C. Li, Y.J. Duan, A new and efficient method for the synthesis of Ulipristal acetate. Steroids 84 (2014) 78–83. DOI:10.1016/j.steroids.2014.03.009 |
| [19] | Y. Zhao, X.L. Li, H. liu, First synthesis and characterization for the stereoisomers of Ulipristal acetate. Steroids 95 (2015) 7–16. DOI:10.1016/j.steroids.2014.12.009 |
| [20] | M. Qian, K. Krishnan, E. Kudova, Neurosteroid analogues. 18. Structure-activity studies of ent-steroid potentiators of γ-aminobutyric acid type A receptors and comparison of their activities with those of alphaxalone and allopregnanolone. J. Med. Chem. 57 (2014) 171–190. DOI:10.1021/jm401577c |
| [21] | J. Roy, P. DeRoy, D. Poirier, 2β-(N-Substituted piperazino)-5α-androstane-3α,17β-diols:parallel solid-phase synthesis and antiproliferative activity on human leukemia HL-60 cells. J. Comb. Chem. 9 (2007) 347–358. DOI:10.1021/cc060098z |
| [22] | C. Varela, E.J. Tavares da Silva, C. Amaral, New structure-activity relationships of A- and D-ring modified steroidal aromatase inhibitors:design, synthesis, and biochemical evaluation. J. Med. Chem. 55 (2012) 3992–4002. DOI:10.1021/jm300262w |
| [23] | J. Roy, R. Maltais, H. Jegham, D. Poirier, Libraries of 2β-(N-Substituted piperazino)-5α-androstane-3α,17β-diols:chemical synthesis and cytotoxic effects on human leukemia HL-60 cells and on normal lymphcytes. Mol. Divers. 15 (2011) 317–339. DOI:10.1007/s11030-010-9273-2 |
| [24] | R. Maltais, A. Hospital, A. Delhomme, J. Roy, D. Poirier, Chemical synthesis, NMR analysis and evaluation on a cancer xenograft model (HL-60) of the aminosteroid derivative RM-133. Steroids 82 (2014) 68–76. DOI:10.1016/j.steroids.2014.01.008 |
| [25] | X. Ke, H. Hu, D. Zhou, X. Hu, A facile and general synthesis of 2β-aminosteroid. Synthesis 8 (2009) 1255–1260. |
| [26] | X. Ke, H. Hu, K. Zhang, X. Hu, Significant steroids:effective and general synthesis of 4α-and 4β-amino-5α-androstanes. Chem. Commun. (2009) 1037–1039. |
| [27] | C. Wang, N.P. Rath, D.F. Covey, Neurosteroid analogues. Part 13:synthesis methods for the preparation of 2β-hydroxygonane derivatives as structural mimics of ent-3α-hydroxysteroid modulators of GABAA receptors. Tetrahedron 63 (2007) 7977–7984. DOI:10.1016/j.tet.2007.05.068 |
| [28] | F. Csende, G. Stájer, Versatlereactivityand catalytic effects of copper(II) halides in organic syntheses. Curr. Org. Chem. 9 (2005) 1737–1755. DOI:10.2174/138527205774610903 |
| [29] | R.M. Mohareb, M.Y. Zaki, N.S. Abbas, Synthesis, anti-inflammatory and antiulcer evaluations of thiazole, thiophene, pyridine and pyran derivatives derived from androstenedione. Steroids 98 (2015) 80–91. DOI:10.1016/j.steroids.2015.03.001 |
| [30] | R.M. Mohareb, F. Al-Omran, R.A. Azzam, Heterocyclic ring extension of estrone:synthesis and cytotoxicity of fused pyran, pyrimidine and thiazole derivatives. Steroids 84 (2014) 46–56. DOI:10.1016/j.steroids.2014.03.012 |
| [31] | R.K. Shiroodi, A.S. Dudnik, V. Gevorgyan, Stereocontrolled 1,3-phosphatyloxy and 1,3-halogen migration relay toward highly functionalized 1,3-dienes. J. Am. Chem. Soc. 134 (2013) 6928–6931. |
| [32] | G.B. Zhang, F.X. Wang, J.Y. Du, Toward the total synthesis of palhinine A:expedient assembly of multifunctionalized isotwistane ring system with contiguous quaternary stereocenters. Org. Lett. 14 (2012) 3696–3699. DOI:10.1021/ol301534r |
| [33] | P. Isabel, M.D. Pawelczyk, K. Hirano, R. Fröhlich, F. Glorius, A family of thiazolium salt derived N-heterocyclic carbenes (NHCs) for organocatalysis:synthesis, investigation and application in cross-benzoin condensation. Eur. J. Org. Chem. 28 (2011) 5475–5484. |
| [34] | B. Sreedhar, P.S. Reddy, M. Madhavi, Rapid and catalyst-free α-halogenation of ketonesusing N-halosuccinamides in DMSO. Synth. Commun. 37 (2007) 4149–4156. DOI:10.1080/00397910701574908 |
| [35] | R.E. Counsell, P.D. Klimstra, 2-Haloandrost-1-ene-3,17-diones, U.S. Patent US2980710(issued April 18, 1961). |
| [36] | I.H. Jung, S.J. Park, S.C. Lee, W.G. Choi, Method for preparing rocuronium bromide, K.R. patent KR2010063370A (issued June 11, 2010). |