 2017, Vol. 28
2017, Vol. 28 



Furan derivatives are valuable structural motifs as an important class of five-membered containing oxygen heterocycles in both naturally occurring and artificial compounds with their diverse and potent biological properties [1-5]. Substituted furans also play an important role in synthetic organic chemistry due to their great potential in material science [6, 7] and their usefulness as building blocks [8-10]. In general, substituted furans were synthesized by the two classical and well-established methods including Feist- Bénary cyclocondensation of 1, 3-dicarbonyl compounds with haloketones and Paal-Knorr cyclocondensation of 1, 4-dicarbonyl compounds [11, 12]. Recently, due to their widespread applications in synthetic pharmaceuticals, agrochemicals, and material science, the development of efficient methods for the preparation of polysubstituted furans has attracted substantial interest from medicinal and synthetic chemists [13-19]. Among them, important protocols involved the transition-metal-catalyzed annulations of allenyl, alkynyl, or cyclopropyl ketones or other derivatives [20-24]. In addition to the conventional annulation synthesis, the functionalization of existing furans was another prominent reaction, such as Pd catalyzed direct arylation of furans via C-H activation [25]. Additionally, a simple ring opening reaction or/and cycloisomerization reaction of donor-acceptor cyclopropanes also has provided powerful means of access to diversely substituted furans [26-31]. Although many traditional methods have proven very effective for the synthesis of substituted furans, efficient approaches for the synthesis of polysubstituted furans with flexible substituent patterns from simple and readily available precursors are of great value.
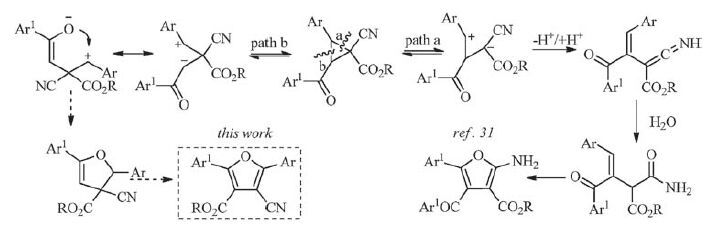
In recent years, D-A cyclopropanes have drawn considerable attention due to the simplicity of their synthesis and potential for variation of the aryl groups, which easily undergo a variety of ringopening reactions under the influence of a variety of conditions [32-36]. A geminal substitution of the donor and acceptor of cyclopropanes induces a high polarization of the C-C bond via the push-pull effect, which can easily be cleaved heterolytically, cyclopropanes are readily transformed into useful 1, 3-dipoles via a ring-opening reaction by treatment with Lewis acids or bases for a multitude of different reactions [37, 38]. As new type of threecarbon synthons, 2-aroyl-3-aryl-1-cyanocyclopropane-1-carboxylates with one donor (aryl) and three acceptor (aroyl, cyano and carboxylate) substituents in the vicinal positions have been used in the construction of aromatic rings. Because of the high polarization of two of three C-C bonds of the ring, the cyclopropanes may be transformed into two 1, 3-dipoles via a ring-opening reaction for the diversity of reactions. Previously, we have reported the opening ring of 1-cyanocyclopropane-1-carboxylates promoted by common organic bases such as piperidine, triethylamine, and DBU [31, 36, 39-41]. Especially, the C-C bond (path a, Scheme 1) of the cyclopropane was cleaved even more easily promoted by combinational iodine/trimethylamine, the ring-opening/cyclization domino reaction of 1-cyanocyclopropane-1-carboxylates formed straightforwardly fully substituted 2-aminofurans [31]. Our results also showed that iodine could promote efficiently these organic transformations as a cost effective, easily available, green, and eco-friendly catalyst [42-44]. Realizing the potential of 1-cyanocyclopropane-1-carboxylates as important building blocks, we embarked in a cycloisomerization strategy for the synthesis of 4-cyanofuran-3-carboxylate derivatives via the cleavage of the C-C bond (path b, Scheme 1) of the cyclopropane.

|
Download:
|
| Scheme 1. Ring-opening/cyclization reaction of 1-cyanocyclopropane-1-carboxylates. | |
Inspired by this study, we herein disclose a novel approach for the construction of fully substituted furans from 1-cyanocyclopropane-1-carboxylates via the iodine/potassium carbonate-mediated ring-opening/cyclization/rearrangement domino reaction (Scheme 1, pathway b). Therefore, the reaction details were carefully investigated and depicted as follows.
2. Results and discussionAt the beginning of our study, the reaction of substrate 1a, prepared easily by treatment of 1- (2- (4-chlorophenyl) -2-oxoethyl) pyridin-1-ium bromide with ethyl 3- (4-chlorophenyl) -2-cyanoacrylate according to reported methods [45, 46], was used as a model reaction for evaluating the effect of the properties of inorganic bases and the amount of iodine on the reaction. The observed results are summarized in Table 1.
|
|
Table 1 Optimization of reaction conditions for the synthesis of 2a.a |

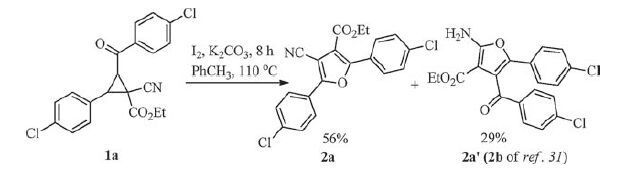
As can be seen, the combinational iodine/sodium hydroxide could not promote efficiently the cycloisomerization reaction after 24 h, just 13% in the yield (entry 1). Pleasingly, a moderate yield (ca. 60%) of the product 4-cyanofuran-3-carboxylate (2a) was obtained using the weak inorganic bases potassium carbonate or cesium carbonate with iodine (entries 2 and 3), the K2CO3/I2 system gave the more acceptable result. Then our efforts further focused on the amount of K2CO3 and I2, the yield was decreased sharply when the amount of K2CO3 was changed from 1.0 equiv. to 0.5 equiv. (Table 1, entry 4) in the presence of 1.0 equiv. iodine for 24 h. While 1.5 equiv. K2CO3 was used under above reaction conditions, the reaction was complete after 12 h and the isolated yield was improved slightly (64%, Table 1, entry 5). When the amount of K2CO3 was increased to 2.0-2.5 equiv., the reaction time was shortened to 8 h and the isolated yields were the best 90% and 89%, respectively (Table 1, entries 6 and 7). Further studies were continued in order to find suitable reaction conditions to accomplish this transformation, we found that, the reaction was carried out under simple conditions that K2CO3 (4 mmol) and substrate 1a (2 mmol) were added in 5 mL of solvent DMF at 110 ℃ for 10 h, a detectable decline in the yield of product 2a appeared when the loading of iodine was decreased from 1.0 equiv. to 0.5 equiv. (Table 1, entry 8). On the contrary, increasing the loading of iodine from 1.0 equiv. to 1.5 equiv. could not markedly accelerate this reaction, however, the reaction time was shortened to 7 h (Table 1, entry 9). Next, based on our previous report [31], toluene as a solvent was investigated for the improvement of the title reaction (Scheme 2), as a result, not only the product 2a was isolated in 56% of yield, but also a side-product 2a' was obtained in 29% of yield, which is identical product in the presence of iodine and trimethylamine using toluene as a solvent. The stronger base K2CO3 promoted mainly the C2-C3 bond cleavage of 2-aroyl-3- aryl-1-cyanocyclopropane-1-carboxylates in comparison with relatively mild and organic base triethylamine. The kinetic intermediate 3-cyano-4-ethoxy-1, 4-dioxo-1-aryl-3- (arylmetheyliumyl) butan-2-ide was obtained easily via stereoselective bond cleavage in the presence of the stronger inorganic base. Contrary to the stronger inorganic base, moderate organic base actually gave a stable and thermodynamics intermediate 3-cyano-4-ethoxy-1, 4- dioxo-1-aryl-3- (arylmetheyliumyl) butan-1-ide (Scheme 1). Additionally, the selective bond cleavage resulted in polar aprotic solvent DMF in comparison with nonpolar aprotic solvent toluene, may also be illustrated by the term of polarizability (Scheme 2).

|
Download:
|
| Scheme 2. The reaction of 1-cyanocyclopropane-1-carboxylate (1a) in toluene | |
These observations clearly indicate that the properties of the inorganic bases and iodine play an important role in this reaction. A series of experiments revealed that the optimal results were obtained when the reaction of 1-cyanocyclopropane-1-carboxylate (1a) together with 1.0 equiv. iodine and 2.0 equiv. K2CO3 was carried out in DMF, the resultant mixture was stirred for 8 h at 110 ℃, whereby the yield of 2a reached 90% (Table 1, entry 6).
With the optimal reaction conditions in hand, we began to explore the substrate scope of 1-cyanocyclopropane-1-carboxylates in this I2/K2CO3 mediated ring-opening/cyclization/rearrangement domino reaction (Table 2). The results indicated that aromatic rings of 1-cyanocyclopropane-1-carboxylates containing electron-withdrawing or electron-donating groups were well tolerated and the desired substituted 4-cyanofuran-3-carboxylates could be afforded in moderate to good yields. In general, aromatic rings of 1-cyanocyclopropane-1-carboxylates bearing electronwithdrawing groups, including para- (1a, 1c, 1e, 1f, 1g), ortho- (1k), and meta- (1j) substituted positions, gave better results. Electrondonating group methoxy (1b, 1d, 1i, 1l) was also well tolerated under the optimized conditions. All corresponding substituted 4-cyanofuran-3-carboxylates were analyzed by their 1H NMR, 13C NMR and MS (Supporting information).
|
|
Table 2 Substrate scope of 1-cyanocyclopropane carboxylates.a |

{kind=link}
{kind=link}
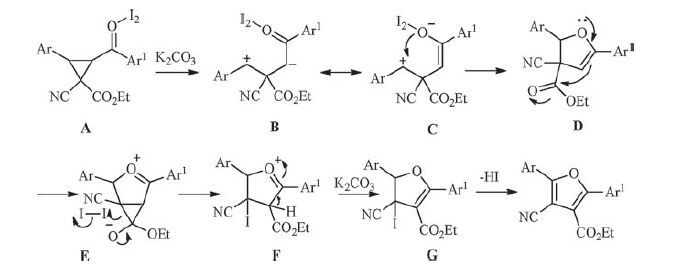
On the basis of the above experimental results together with related reports, presumably the reaction seems to occur following the mechanistic pathway demonstrated in Scheme 3. First of all iodine polarized and activated the carbonyl group of arylketone [44] as it behaved as a mild Lewis acid [47]. In the first step iodine binded with the carbonyl group of arylketone to give an iodine- carbonyl complex [A] and thus increased the withdrawingelectron character of the carbonyl carbon of arylketone [48]. Then potassium carbonate promoted selectively the cleavage of the C-C bond (path b, Scheme 1) of the cyclopropane to form 3-cyano-4- ethoxy-1, 4-dioxo-1-aryl-3- (arylmetheyliumyl) butan-2-ide [B], Next, the tautomerism of butan-2-ide [B] formed an intermediate 2-cyano-2- (ethoxycarbonyl) - 1, 4-diarylbut-3-en-1-ylium-4-olate [C], which underwent an intramolecularly nuclophilic coupling on phenylmethylium ion to give a dihydrofuran intermediate [D]. The dihydrofuran intermediate [D] was transformed to the bicyclic cyclopropane[b]furan intermediate [E] via an intramolecular nucleophilic addition to the carbonyl group of ester again. Further the iodination at α-carbon of cyano group together with ring opening of cyclopropane and following ethoxycarbonyl-transfer reaction formed an intermediate 3-cyano-4- (ethoxycarbonyl) -3- iodo-2, 5-diaryl-3, 4-dihydro-2H-furan-1-ium [F]. In the presence of potassium carbonate the tautomerism of the an iododihydrofuran [F] formed ethyl 4-cyano-4-iodo-2, 5-diaryl-4, 5-dihydrofuran-3-carboxylate intermediate [G]. Finally, removal of HI and aromatization of the iododihydrofuran [G] resulted in the formation of the corresponding 4-cyano-3-furancarboxylates under basic conditions.

|
Download:
|
| Scheme 3. Possible mechanism in the synthesis of 4-cyanofuran-3-carboxylates. | |
{kind=link}
3. Conclusion
In conclusion, we have developed a straightforward and efficient approach for the synthesis of alkyl 4-cyanofuran-3- carboxylates via iodine/potassium carbonate-promoted ringopening/cyclization/rearrangement domino reaction of 1-cyanocyclopropane-1-carboxylates. The operational simplicity, ready availability of reactants, and good chemical yields make this novel synthetic method appealing in diversity-oriented synthesis. This protocol is expected to find considerable applications in the synthesis of polysubstituted furancarboxylates, which are a wide range of structurally interesting and pharmacologically significant compounds.
4. ExperimentalAll melting points were determined in a Yanaco melting point apparatus and are uncorrected. IR spectra were recorded in a Nicolet FT-IR 5DX spectrometer. The 1H NMR (400 or 600 MHz) and 13C NMR (100 or 150 MHz) spectra were recorded in a Bruker AV-600 spectrometer with TMS as internal reference in CDCl3 solutions. The J values are given in hertz. Only discrete or characteristic signals for the 1H NMR are reported. The MS spectra were obtained on a ZAB-HS mass spectrometer with 70 eV. Flash chromatography was performed on silica gel (230-400 mesh) eluting with ethyl acetate-hexanes mixture. All reactions were monitored by thin layer chromatography (TLC). All reagents and solvents were purchased from commercial sources and purified commonly before used.
4.1. General procedure for preparation of 2, 5-diaryl-4-cyanofuran-3- carboxylates (2a-l)The appropriate 2-aroyl-3-aryl-1-cyanocyclopropanecarboxylates (2 mmol), potassium carbonate (552 mg, 4 mmol) and iodine (510 mg, 2 mmol) were dissolved in 5 mL of DMF, and the resultant mixture was stirred at room temperature for 15 min. The reaction mixture was stirred at 110 ℃ for 8 h, and the completion of reaction was confirmed by TLC (EtOAc/hexanes 1:15). Subsequently, the solvent was removed under reduced pressure, to the residues was added with water (10 mL) followed by extraction with dichloromethane (10 mL × 2). The organic phase was washed with water (15 mL) and brine (10 mL), and dried over anhydrous sodium sulfate. After removal of dichloromethane, the crude product was purified by flash chromatography (silica gel, EtOAc/hexanes, 1/50) to give the desired 2a-l.
Ethyl 2, 5-bis (4-chlorophenyl) -4-cyanofuran-3-carboxylate (2a) : White solid, 90%; mp 171.2-171.9 ℃ (EA/PE); IR (KBr, cm-1) : v 3433, 3106, 2981, 2936, 2232, 1712, 1594, 1483, 1401, 1368, 1327, 1222, 1157, 1103, 1017, 969, 833, 785, 711, 672, 483; 1H NMR (400 MHz, CDCl3) : δ 7.92 (d, 2H, J = 8.8 Hz), 7.86 (d, 2H, J = 8.8 Hz), 7.40 (d, 2H, J = 8.8 Hz), 7.38 (d, 2H, J = 8.8 Hz), 4.33 (q, 2H, J = 7.2 Hz), 1.34 (t, 3H, J = 7.2 Hz); 13C NMR (CDCl3, 100 MHz) : δ 160.9, 157.9, 155.9, 137.0, 136.8, 130.0, 129.5, 128.7, 127.0, 126.1, 125.5, 115.2, 113.5, 94.4, 61.9, 13.9; MS (EI) (m/z) : 408.02 [(M+Na)+] (100%).
AcknowledgmentsFinancial support of this research by the National Natural Science Foundation of China (NSFC No. 21173181) is gratefully acknowledged by authors. A Project was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.09.017.
| [1] | A. Corma, S. Iborra, A. Velty, Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 107 (2007) 2411–2502. DOI:10.1021/cr050989d |
| [2] | B.H. Lipshutz, Five-membered heteroaromatic rings as intermediates in organic synthesist. Chem. Rev. 86 (1986) 795–891. DOI:10.1021/cr00075a005 |
| [3] | A.U. Rao, D. Xiao, X. Huang, Facile synthesis of tetracyclic azepine and oxazocine derivatives and their potential as MAPKAP-K2(MK2) inhibitors. Bioorg. Med. Chem. Lett. 22 (2012) 1068–1072. DOI:10.1016/j.bmcl.2011.11.113 |
| [4] | N. Kumari, C.B. Mishra, A. Prakash, 8-(Furan-2-yl)-3-phenethylthiazolo[5,4-e] [1,2,4] triazolo[1,5-c]pyrimidine-2(3H)-thione as novel, selective and potent adenosine A2A receptor antagonist. Neurosci. Lett. 558 (2014) 203–207. DOI:10.1016/j.neulet.2013.10.035 |
| [5] | F. Hasegawa, K. Niidome, C. Migihashi, Discovery of furan-2-carbohydrazides as orally active glucagon receptor antagonists. Bioorg. Med. Chem. Lett. 24 (2014) 4266–4270. DOI:10.1016/j.bmcl.2014.07.025 |
| [6] | O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov, D.F. Perepichka, Towards green electronic materials. α-Oligofurans as semiconductors. Chem. Commun. 47 (2011) 1976–1978. DOI:10.1039/C0CC04699J |
| [7] | C. Zeng, H. Seino, J. Ren, K. Hatanaka, N. Yoshie, Bio-based furan polymers with self-healing ability. Macromolecules 46 (2013) 1794–1802. DOI:10.1021/ma3023603 |
| [8] | M.M. Abd-Elzaher, A.A. Labib, H.A. Mousa, S.A. Moustafa, M.M. Abdallah, Synthesis, characterization and cytotoxic activity of ferrocenyl hydrazone complexes containing a furan moiety. Res. Chem. Intermed. 40 (2014) 1923–1936. DOI:10.1007/s11164-013-1090-7 |
| [9] | Z.L. Wang, H.L. Li, L.S. Ge, DDQ-mediated oxidative coupling:an approach to 2,3-dicyanofuran (thiophene). J. Org. Chem. 79 (2014) 1156–1165. DOI:10.1021/jo4026034 |
| [10] | D. Kalaitzakis, M. Triantafyllakis, I. Alexopoulou, M. Sofiadis, G. Vassilikogiannakis, One-pot transformation of simple furans into 4-hydroxy-2-cyclopentenones in water. Angew. Chem. Int. Ed. 53 (2014) 13201–13205. DOI:10.1002/anie.201407477 |
| [11] | G. Mross, E. Holtz, P. Langer, Synthesis of 2-alkenyl-3-(alkoxycarbonyl)furans based on feist-benary cyclocondensation of (2,4-dioxobutylidene) phosphoranes with α-haloketones and α-chloracetaldehyde. J. Org. Chem. 71 (2006) 8045–8049. DOI:10.1021/jo061153t |
| [12] | G. Minetto, L.F. Raveglia, A. Sega, M. Taddei, Microwave-assisted Paal-Knorr reaction-three-step regiocontrolled synthesis of polysubstituted furans, pyrroles and thiophenes. Eur. J. Org. Chem. (2005) 5277–5288. |
| [13] | B.A. Keay, Synthesis of multi-substituted furan rings:the role of silicon. Chem. Soc. Rev. 28 (1999) 209–215. DOI:10.1039/a809439j |
| [14] | M. Miao, X. Xu, L. Xu, H. Ren, Copper(I) iodide mediated iodocyclization of cyclopropylideneallenyl ketones:facile and effective synthesis of highly substituted furan derivatives. Eur. J. Org. Chem. (2014) 5896–5900. |
| [15] | J. Daru, Z. Benda, Á. Po'ti, Z. Nova'k, A. Stirling, Mechanistic study of silvermediated furan formation by oxidative coupling. Chem. Eur. J. 20 (2014) 15395–15400. DOI:10.1002/chem.v20.47 |
| [16] | Y. Yang, J. Yao, Y. Zhang, Synthesis of polysubstituted furans via coppermediated annulation of alkyl ketones with α,β-unsaturated carboxylic acids. Org. Lett. 15 (2013) 3206–3209. DOI:10.1021/ol400912v |
| [17] | C. He, S. Guo, J. Ke, Silver-mediated oxidative C-H/C-H functionalization:a strategy to construct polysubstituted furans. J. Am. Chem. Soc. 134 (2012) 5766–5769. DOI:10.1021/ja301153k |
| [18] | P. Lenden, D.A. Entwistle, M.C. Willis, An alkyne hydroacylation route to highly substituted furans. Angew. Chem. Int. Ed. 50 (2011) 10657–10660. DOI:10.1002/anie.201105795 |
| [19] | J. Wu, N. Yoshikai, Modular synthesis of multisubstituted furans through palladium-catalyzed three-component condensation of alkynylbenziodoxoles carboxylic acids, and imines. Angew. Chem. Int. Ed. 54 (2015) 11107–11111. DOI:10.1002/anie.201504687 |
| [20] | S. Ma, Transition metal-catalyzed/mediated reaction of allenes with a nucleophilic functionality connected to the α-carbon atom. Acc. Chem. Res. 36 (2003) 701–712. DOI:10.1021/ar020133k |
| [21] | Z.P. Zhan, S.P. Wang, X.B. Cai, Copper(II) triflate-catalyzed nucleophilic substitution of propargylic acetates with enoxysilanes. A straightforward synthetic route to polysubstituted furans. Adv. Synth. Catal. 349 (2007) 2097–2102. DOI:10.1002/(ISSN)1615-4169 |
| [22] | M.L. Hossain, F. Ye, Y. Zhang, J. Wang, Cu(I)-catalyzed reaction of diazo compounds with terminal alkynes:a direct synthesis of trisubstituted furans. Tetrahedron 70 (2014) 6957–6962. DOI:10.1016/j.tet.2014.07.086 |
| [23] | T.J. Donohoe, J.F. Bower, An expedient route to substituted furans via olefin cross-metathesis. Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 3373–3376. DOI:10.1073/pnas.0913466107 |
| [24] | M. Zhang, H.F. Jiang, H. Neumann, M. Beller, P.H. Dixneuf, Sequential synthesis of furans from alkynes:successive ruthenium(II)-and copper(II)-catalyzed processes. Angew. Chem. Int. Ed. 48 (2009) 1681–1684. DOI:10.1002/anie.200805531 |
| [25] | A. Battace, M. Lemhadri, T. Zair, H. Doucet, M. Santelli, Palladium-catalyzed direct arylation of furans via C-H functionalization at low catalyst loadings. Organometallics 26 (2007) 472–474. DOI:10.1021/om0610243 |
| [26] | A.T. Parsons, J.S. Johnson, Catalytic enantioselective synthesis of tetrahydrofurans:a dynamic kinetic asymmetric 3+2 cycloaddition of racemic cyclopropanes and aldehydes. J. Am. Chem. Soc. 131 (2009) 3122–3123. DOI:10.1021/ja809873u |
| [27] | Y. Miyake, S. Endo, T. Moriyama, K. Sakata, Y. Nishibayashi, Rutheniumtriggered ring opening of ethynylcyclopropanes:[3+2] cycloaddition with aldehydes and aldimines involving metal allenylidene intermediates. Angew. Chem. Int. Ed. 52 (2013) 1758–1762. DOI:10.1002/anie.v52.6 |
| [28] | G. Yang, Y. Sun, Y. Shen, cis-2,3-Disubstituted cyclopropane 1,1-diesters in[3+2] annulations with aldehydes:highly diastereoselective construction of densely substituted tetrahydrofurans. J. Org. Chem. 78 (2013) 5393–5400. DOI:10.1021/jo400554a |
| [29] | C. Zhang, M. Xu, J. Ren, Z. Wang, Sc(OTf)3-catalyzed diastereoselective formal[3+2] cycloaddition reactions of alkynylcyclopropane ketones with electronrich aromatic aldehydes to yield 2,5-trans-tetrahydrofurans. Eur. J. Org. Chem. (2016) 2467–2478. |
| [30] | Y. Zhu, P. Xu, Y. Gong, Triflic acid-catalyzed cycloisomerization reactions of donor-acceptor cyclopropanes:access to alkyl 5-arylfuran-2-carboxylates. J. Org. Chem. 81 (2016) 4829–4834. DOI:10.1021/acs.joc.6b00161 |
| [31] | W. Ye, C. Tan, J. Yao, S. Xue, Y. Li, C. Wang, Iodine-promoted domino reactions of 1-cyanocyclopropane 1-esters:a straightforward approach to fully substituted 2-aminofurans. Adv. Synth. Catal. 358 (2016) 426–434. DOI:10.1002/adsc.201500078 |
| [32] | M.A. Cavitt, L.H. Phun, S. France, Intramolecular donor-acceptor cyclopropane ring-opening cyclizations. Chem. Soc. Rev. 43 (2014) 804–818. DOI:10.1039/C3CS60238A |
| [33] | T.F. Schneider, J. Kaschel, D.B. Werz, A new golden age for donor-acceptor cyclopropanes. Angew. Chem. Int. Ed. 53 (2014) 5504–5523. DOI:10.1002/anie.v53.22 |
| [34] | H.K. Grover, M.R. Emmett, M.A. Kerr, Carbocycles from donor-acceptor cyclopropanes. Org. Biomol. Chem. 13 (2015) 655–671. DOI:10.1039/C4OB02117G |
| [35] | D.J.N. Mack, J.T. Njardarson, Recent advances in the metal-catalyzed ring expansions of three-and four-membered rings. ACS Catal. 3 (2013) 272–280. DOI:10.1021/cs300771d |
| [36] | J. Liu, L. Zhou, W. Ye, C. Wang, Formal[3+2] cycloaddition of 1-cyanocyclopropane 1-ester with pyridine, quinoline or isoquinoline:a general and efficient strategy for construction of cyanoindolizine skeletons. Chem. Commun. 50 (2014) 9068–9071. DOI:10.1039/C4CC03267E |
| [37] | M.Y. Mel'nikov, E.M. Budynina, O.A. Ivanova, I.V. Trushkov, Recent advances in ring-forming reactions of donor-acceptor cyclopropanes. Mendeleev Commun. 21 (2011) 293–301. DOI:10.1016/j.mencom.2011.11.001 |
| [38] | Z. Wang, Polar intramolecular cross-cycloadditions of cyclopropanes toward natural product synthesis. Synlett 23 (2012) 2311–2327. DOI:10.1055/s-00000083 |
| [39] | C. Tan, W. Ye, J. Yao, Efficient strategy for construction of 6-carbamoylfulvene-6-carboxylate skeletons via[3+2] cycloaddition of 1-cyanocyclopropane 1-ester with β-nitrostyrenes. RSC Adv. 5 (2015) 26491–26497. DOI:10.1039/C5RA02918J |
| [40] | W. Ye, L. Zhou, S. Xue, Y. Li, C. Wang, Bimolecular intermolecular-Michael/intramolecular-Michael/aromatization reaction of 1-cyanocyclopropane 1-esters or 1,1-dicyanocyclopropanes:a straightforward approach to fully substituted benzenes. Synlett 26 (2015) 1769–1777. DOI:10.1055/s-00000083 |
| [41] | S. Xue, J. Liu, C. Wang, DBU-mediated[3+2] cycloaddition reactions of donor-acceptor cyclopropanes with nitromethane:efficient strategy for the construction of isoxazole skeletons. Eur. J. Org. Chem. (2016) 2450–2456. |
| [42] | P. Rai, M. Srivastava, J. Singh, J. Singh, Molecular iodine:a green and inclusive catalyst for the synthesis of highly functionalized 1,3,5-trisubstituted pyrazoles in aqueous medium. RSC Adv. 4 (2014) 779–783. DOI:10.1039/C3RA44315A |
| [43] | H. Song, Y. Liu, Q. Wang, Cascade electrophilic iodocyclization:efficient preparation of 4-iodomethyl substituted tetrahydro-β-carbolines and formal synthesis of oxopropaline G. Org. Lett. 15 (2013) 3274–3277. DOI:10.1021/ol401303f |
| [44] | M. Kidwai, V. Bansal, P. Mothsra, One-pot synthesis of highly substituted imidazoles using molecular iodine:a versatile catalyst. J. Mol. Catal. A:Chem. 265 (2007) 177–182. DOI:10.1016/j.molcata.2006.10.009 |
| [45] | A.E. Raveendran, R.R. Paul, E.S.V. Nair, Nucleophilic heterocyclic carbene as a novel catalyst for cyclopropanation of cyano acrylates. Org. Biomol. Chem. 8 (2010) 901–905. DOI:10.1039/B916343C |
| [46] | V.R.K. Kanagaraj, T. Subramanian, P. Suresh, K. Pitchumani, Pyridinium ylideassisted KY zeolite catalyzed tandem synthesis of polysubstituted cyclopropanes. Catal. Commun. 26 (2012) 39–43. DOI:10.1016/j.catcom.2012.04.020 |
| [47] | J. Sun, Y. Dong, X. Wang, S. Wang, Y. Hu, Highly efficient chemoselective deprotection of o,o-acetals and o,o-ketals catalyzed by molecular iodine in acetone. J. Org. Chem. 69 (2004) 8932–8934. DOI:10.1021/jo0486239 |
| [48] | M. Kidwai, V. Bansal, P. Mothsra, Molecular iodine:a versatile catalyst for the synthesis of bis(4-hydroxycoumarin) methanes in water. J. Mol. Catal. A:Chem. 268 (2007) 76–81. DOI:10.1016/j.molcata.2006.11.054 |