 2017, Vol. 28
2017, Vol. 28 


 , Zhen-Yuan Miaob, Wan-Nian Zhanga,b
, Zhen-Yuan Miaob, Wan-Nian Zhanga,b
b School of Pharmacy, Second Military Medical University, Shanghai 200433, China
The tumor suppressor p53 plays a vital role in human cancers. The overexpression of MDM2, a negative regulator protein of p53, has been shown in many human cancer cells [1, 2]. The new cancer therapeutic strategy to activate p53 releasing from MDM2 has been investigated and the first small molecule inhibitor Nultin-3 was identified in 2004 [3]. To date, many approaches have been described and a series of small molecule inhibitors with excellent antitumor activities have been reported [4-9]. Currently, seven MDM2 inhibitors have advanced into clinical trials for the treatment of neoplasms, myeloma and acute myeloid leukemia [10]. Among them, RG7112, RG7338, MI77301, NVP-CGM097 and AMG232 have been disclosed the structures with their clinical data in detail (Fig. 1).

|
Download:
|
| Figure 1. The reported structures of p53-MDM2 inhibitors in clinical trials. | |
In our previous work, pyrrolo[3-4c]pyrazol-6 (1H) -ones have been discovered by structure-based virtual screening method as potent p53-MDM2 inhibitors [11]. Further mechanistic study indicated these compounds simultaneously inhibited p53-MDM2 interaction and the NF-κB pathway [12, 13]. This scaffold was predicted to mimic the p53 peptide and insert into the three hotspots (namely Phe19, Trp23, and Leu26) of MDM2 protein [13]. The previous study only focused on the position N1 of pyrroloc[3-4c]pyrazol-6 (1H) -one scaffold that located in Phe19 pocket. The other two phenyl groups that are also key features of the scaffold inserting into the Trp23 and Leu26 hydrophobic pockets have not been explored. In this paper, we described the relevant continuous optimization and structure-activity relationship of 4, 5-dihydropyrrolo[3-4c]pyrazol-6 (1H) -ones for anticancer drug development.
2. Results and discussion 2.1. ChemistryThe target compounds were synthesized in four steps using acetophenone and 1- (4-chlorophenyl) ethan-1-one as starting materials (Scheme 1). Firstly, 1a or 1b was condensed with ethyl oxalate in methanol to afford 2a or 2b. Then, pyrazol-6 (1H) -ones 3a-3f were obtained in a three-component coupling reaction in good yields. Compounds 3a-3f were subsequently condensed with excessive hydrazine and then substituted with various aryl groups to give novel 4, 5-dihydropyrrolo [3, 4-c]pyrazole derivatives 5a-5n.
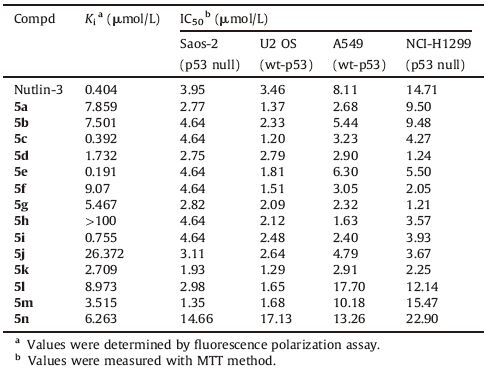
2.2. p53-MDM2 protein-protein interaction inhibitory activityThe fluorescent polarization assay indicated that most compounds showed potent binding activities to MDM2 except compounds 5h and 5j (Table 1). Overall, mono-substitution on the phenyl group (5c-5e) exhibited better inhibitory activities than the di-substituted ones (5g and 5h). To our delight, compounds 5c, 5e and 5i possessed significant Ki values of 0.392 μmol/L, 0.191 μmol/L, and 0.755 μmol/L, respectively. Especially, the former two compounds exhibited the increased Ki values compared with the positive drug Nultin-3 (Ki = 0.404 μmol/L). Structure- activity relationship (SAR) analysis demonstrated that compounds with benzyl groups (N1-3'-methylbenzyl 5c and N1-4'-fluorobenzyl 5e) on the position N1 of pyrazole scaffold showed higher MDM2 binding activities than alkyl groups (N1-cyclopentyl 5d and N1- pentyl 5f). The result was further confirmed by introduction of the N1-pentyl group on the pyrazole ring with the negative effects of compounds 5h and 5j. Differently, with none substitution on the basic benzyl group, the compound (5l) was less potent than that with N1-pentyl (5k). Cyclic alkyl substitution (5d) on N1 position was much active than the corresponding alkane (5f). Among the halogen substitutions of fluorine, chloride and bromine on position of C4-phenyl, bromine derivatives indicated good binding activities. Bromine-compounds (5c-5e) showed better inhibitory activities than fluorine substituted compounds (5a and 5b). Di-substituted groups (5g and 5h) and non-substitution (5k-5m) on the benzene ring were unfavorable to the activities. In order to confirm the binding activities of compound 5e, the docking experiment was performed. To our delight, the compound well mimicked the p53 key features. Except the N1-benzyl group, the other three aromatic rings insert into the Phe19, Trp23, and Leu26 hydrophobic pockets of MDM2 protein, respectively ( Fig. S1 in Supporting information), which matched well with the original ligand benzodiazepine of the crystal complex (PDB #1T4E). The N1-4-fluorobenzyl group was located at the solvent exposed site of the MDM2 protein.
|
|
Table 1 In vitro antiproliferative and p53-MDM2 inhibitory activities of compounds. |
{kind=link}

|
Download:
|
| Scheme1. The synthetic route of 4, 5-dihydropyrrolo[3, 4-c]pyrazol-6 (1H) -ones. Reagents and conditions: (a) ethyl oxalate, CH3ONa, CH3OH, 60 ℃, 2 h; (b) benzaldehyde derivatives, N- (3-aminopropyl) -imidazole, 1, 4-dioxane, r.t., 12 h, 65% for two steps; (c) 80% N2H4·H2O, CH3COOH, 0 ℃ to reflux, 8 h, 82%; (d) RX, K2CO3, DMF, 50 ℃, 4 h, 28.5- 70.1%. | |
{kind=link}
2.3. Antiproliferative activities and western blotting assay
Two cancer cell lines with wild type p53 (U2 OS and A549) and the corresponding cell lines with deficient p53 (Saos-2 and NCIH1299) were selected to evaluate the antiproliferative activities of new pyrrolo[3, 4-c]pyrazol-6 (1H) -ones. As shown in Table 1, it is readily to find that fourteen compounds showed potent activities against all cancer lines. For the human lung cancer A549, eleven compounds possessed higher activities than Nutlin-3. Among these compounds, 5a, 5g, 5h and 5i exhibited 3-fold increased activities than the positive drug. Different to the result of A549 cells, compounds 5d and 5g showed 12-fold higher activities than Nultin- 3 against NCI-H1299. For the human sarcoma, target compounds indicated potent activities against U2 OS cells except 5n. N1-30- methylbenzyl substituted compound 5c showed the excellent antiproliferative activity with an IC50 value of 1.20 mmol/L, which is 3-fold stronger than that of Nultin-3. In contrast, introducing the hydroxypropyl group (5n) resulted in the sharply decrease in the activities. Furthermore, compound 5a, 5b and 5c indicated good selectivity for both lung cancer and sarcoma cells. For instance, compound 5c showed 4-fold selectivity of U2 OS and Saos-2 while slightly selectivity of Nultin-3 was demonstrated. In addition, compounds 5e, 5f, 5h and 5i showed 2-3-fold selection in sarcoma cells.
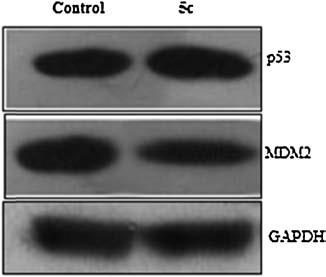
To further validate the mechanism of compound 5c, western blotting was used for analyzing the levels of p53 and MDM2 proteins in A549 cancer cells. Consistent with the result of the fluorescence polarization assay and our previous results [11-13], 1 μmol/L of compound 5c could increase p53 protein and decrease MDM2 protein after 4 h treatment (Fig. 2; all the gels were from the same experiment), mechanistically supporting this class of compound inhibiting p53-MDM2 interaction.

|
Download:
|
| Figure 2. Cellular activity of compound 5c for the p53 pathway activation detected by Western blotting assay (A549 cells, 1 mmol/L, 4 h treatment). | |
{kind=link}
3. Conclusion
In summary, the SAR study revealed the contribution of benzyl group on position N1 to the binding activity. Compound 5c with N1-30-methylbenzyl and compound 5e with N1-40-fluorobenzyl showed increased activities compared with Nultin-3. In addition, among the halo-substitution on position C4 of the phenyl, bromine exhibited higher activity than fluorine and chloride. Optimization of 4, 5-dihydropyrrolo[3, 4-c]pyrazol-6 (1H) -ones also led to discover the potent p53-MDM2 inhibitor 5c with 4-fold selectivity for wt-p53 cancer cells (U2 OS) and p53-deleted cancer cells (Saos-2). The present findings might been valuable for the further development of pyrrolo[3, 4-c]pyrazol-6 (1H) -ones as potent p53- MDM2 inhibitors.
4. Experimental 4.1. ChemistryThe 1H NMR and 13C NMR spectra were recorded at 300 or 600 MHz with a Bruker instrument, and reported with TMS as internal standard and CDCl3 or DMSO-d6 as solvents. Chemical shifts (δ values) and coupling constants (J values) are given in ppm and Hz, respectively. ESI mass spectrometry was performed on an API-3000 LC-MS spectrometer. TLC analysis was carried out on silica gel plates GF254 (Qindao Haiyang Chemical, China). Flash column chromatography was carried out on silica gel 300-400 mesh. Anhydrous solvent and reagents were all analytical pure and dried through routine protocols.
Synthesis of 5- (3- (1H-imidazole-1-yl) propyl) -3- (4-chorophenyl) -4- (4-fluorophenyl) -1- (4-trifluoromethylbenzyl) -4, 5-dihydropyrrolo[3, 4-c]pyrazol-6 (1H) -one (5a) : Compound 4e (0.21 g, 0.5 mmol), 4-trifulorobenzyl bromide (0.12 g, 1.0 mmol) and potassium carbonate (0.14 g, 1.0 mmol) were mixtured in DMF (10 mL) and stirred at 50 ℃ for 4 h. After cooling to room temperature, the solution mixture was added to water (50 mL) and then extracted with EtOAc. The organic layer was then washed with brine, dried (Na2SO4), and evaporated to dryness. The residue was purified by flash column chromatography (CH2Cl2:CH3OH = 100:2) to give compound 5a, yield: 45%, m.p. <50 ℃. 1H NMR (300 MHz, DMSO-d6) : δ 1.78-1.93 (m, 2 H), 2.60- 2.69 (m, 1 H), 3.87-3.96 (m, 2 H), 3.96-4.05 (m, 1H), 5.64 (s, 2H), 6.00 (s, 1H), 6.85 (s, 1H), 7.10-7.20 (m, 3H), 7.30-7.35 (m, 4H), 7.46 (d, 2H, J = 8.17 Hz), 7.60 (d, 3H, J = 7.21 Hz), 7.77 (d, 2H, J = 8.38 Hz). 13C NMR (75 MHz, DMSO-d6) : δ 142.76, 141.58, 141.17, 137.74, 133.26, 131.86, 131.82, 130.82, 130.75, 130.63, 130.42, 129.16, 129.03, 128.72, 127.93, 126.19, 119.69, 116.62, 116.34, 60.26, 58.25, 53.21, 43.98, 38.10, 31.39, 29.78, 22.53, 21.27, 14.53, 14.42. MS (ESI) : m/z [M+H]+: 594.36.
General procedure and characterization data for the preparation of compounds 5b-5n and corresponding intermediates are detailed in the Supporting information.
4.2. Fluorescence polarization binding assayBriefly, the fluorescence polarization experiments were read on Biotek Synergy H2 with the 485 nm excitation and 535 nm emission filters. The fluorescence intensities parallel (Intparallel) and perpendicular (Intperpedicular) to the plane of excitation were measured in black 96-well plates with nonbinding surface (Corning #3993) at room temperature. The background fluorescence intensities of blank samples containing the reference buffer were subtracted, and steadystate fluorescence polarization was calculated using the following equation: P = 1000 × (Intparallel - GIntperpedicular) / (Intparallel + GIntperpedicular), and the correction factor G was determined by standard polarization of fluorescein in order to eliminate differences in the transmission of vertically and horizontally polarized light. All fluorescence polarization values were expressed in millipolarization units (mP). The dosedependent binding experiments were carried out with serial dilution in DMSO of compounds. A 5 μL sample of the test sample, preincubated (for 30 min) MDM2 binding domain (1-118) (10 nmol/L), and PMDM6-F peptide (Anaspec) (10 nmol/L) in assay buffer (100 mmol/L potassium phosphate, pH 7.5; 100 mg/mL bovine gamma globulin; 0.02% sodium azide) were added to microplates to produce a final volume of 115 mL. For each assay, the controls contained the MDM2 binding domain and PMDM6-F. Plates were read at 1 h after mixing all assay components. Binding constant (Ki) was determined by fitting inhibition curves using GraphPad Prism software. Nutlin- 3a (Sigma-Aldrich) was used as reference compound for validating the assay in each plate.
4.3. In vitro antiproliferative assayCells were plated in 96-well plates at a density of 5 × 103/ well and incubated in a humidified atmosphere with 5% CO2 at 37 ℃ for 24 h. Compounds 5a-5n were added onto triplicate wells with different concentrations and 0.1% DMSO for control. After incubated for 72 h, 20 mL of MTT (3- (4, 5-dimethyl thiazol- 2-yl) -2, 5-diphenyltetrazolium bromide) solution (5 mg/mL) was added to each well and the plate was incubated for an additional 4 h. The formazan was dissolved in 100 μL of DMSO. The absorbance (OD) was read on a WellscanMK-2 microplate reader (Labsystems) at 570 nm. The concentration causing 50% inhibition of cell growth (IC50) was determined by the Logit method.
The western blotting assay and docking experiment were followed our previous method [11-13].
AcknowledgmentsThis work was supported in part by the National Natural Science Foundation of China (Nos. 81373331 and 81502978), the Bio-Pharmaceutical Project of Science and Technology of Shanghai (No. 14431902300) and Youth grant of Second Military Medical University (No. 2014QN08).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.09.001.
| [1] | N.J. Stephen, R.H. Amy, V. Hannes, Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. U. S. A. 95 (1998) 15608–15612. DOI:10.1073/pnas.95.26.15608 |
| [2] | E. Rayburn, R. Zhang, J. He, H. Wang, MDM2 and human malignancies:expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 5 (2005) 27–41. DOI:10.2174/1568009053332636 |
| [3] | L.T. Vassilev, B.T. Vu, B. Graves, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303 (2004) 844–848. DOI:10.1126/science.1092472 |
| [4] | S. Wang, W. Sun, Y. Zhao, SAR405838:an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 74 (2014) 5855–5865. DOI:10.1158/0008-5472.CAN-14-0799 |
| [5] | B. Vu, P. Wovkulich, G. Pizzolato, Discovery of RG7112:a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4 (2013) 466–469. DOI:10.1021/ml4000657 |
| [6] | Q. Ding, Z. Zhang, J.J. Liu, Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 56 (2013) 5979–5983. DOI:10.1021/jm400487c |
| [7] | Y. Rew, D. Sun, Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J. Med. Chem. 57 (2014) 6332–6341. DOI:10.1021/jm500627s |
| [8] | P. Holzer, K. Masuya, P. Furet, Discovery of a dihydroisoquinolinone derivative (NVP-CGM097):a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J. Med. Chem. 58 (2015) 6348–6358. DOI:10.1021/acs.jmedchem.5b00810 |
| [9] | Z. Zhang, Q. Ding, J.J. Liu, Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorg. Med. Chem. 22 (2014) 4001–4009. DOI:10.1016/j.bmc.2014.05.072 |
| [10] | Y. Zhao, A. Aguilar, D. Bernard, Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 58 (2015) 1038–1052. DOI:10.1021/jm501092z |
| [11] | C. Zhuang, Z. Miao, L. Zhu, Discovery, synthesis and biological evaluation of orally active pyrrolidone derivatives as novel Inhibitors of p53-MDM2 protein-protein interaction. J. Med. Chem. 55 (2012) 9630–9642. DOI:10.1021/jm300969t |
| [12] | C. Zhuang, Z. Miao, Y. Wu, Double-edged swords as cancer therapeutics:novel, orally active, small molecules simultaneously inhibit p5-MDM2 interaction and the NF-кB pathway. J. Med. Chem. 57 (2014) 567–577. DOI:10.1021/jm401800k |
| [13] | C. Zhuang, C. Sheng, W.S. Shin, A novel drug discovery strategy:mechanistic investigation of an enantiomeric antitumor agent targeting dual p53 and NF-кB pathways. Oncotarget 21 (2014) 10830–10839. |