 2017, Vol. 28
2017, Vol. 28 


 , Yan-Ping Chenb, Yong Shenb
, Yan-Ping Chenb, Yong Shenb
b College of Agriculture and Biotechnology, Yunnan Agricultural University, Kunming 650201, China;
c State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China
Aconitum nagarum var. heterotrichum, called 'xiao bai cheng' in Chinese, is a variant of A. nagarum. This variety is distinguished from the typical one with the indumentum of dense spreading yellowish glandular hairs on both the inflorescence axis and the pedicel, mainly distributing in Dengchuan, Gongshan and Qiaojia etc. in Yunnan Province . Its roots are highly toxic, and also can be used for treating lumbar muscle strain, rheumatism and other diseases by natives [2]. Previous phytochemical investigations of this plant revealed that C19-diterpenoid alkaloids were the primary constituents [3-5]. As a part of an ongoing phytochemical investigation of A. nagarum var. heterotrichum, four new diterpenoid alkaloids, named nagaconitines A-D (1-4, Fig. 1), were isolated from the roots of this medicinal plant. Moreover, their activities against cancer cell line SK-OV-3 were measured in vitro, and compounds 3-4 showed antitumor effect. This paper described their isolation, structural elucidation and antitumor results.

|
Download:
|
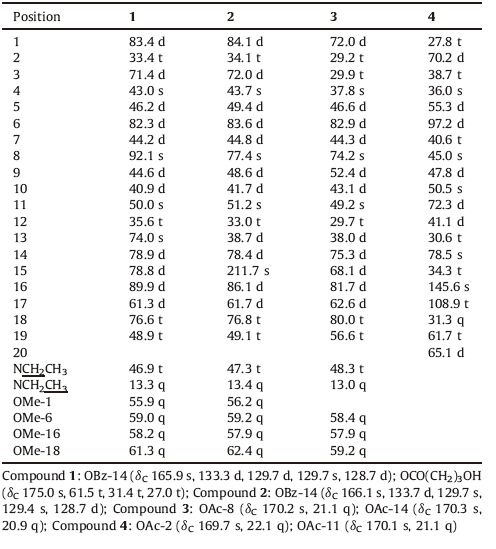
| Figure 1. The structures of compounds 1-4, isolated from Aconitum nagarum var. heterotrichum. | |
2. Results and discussion
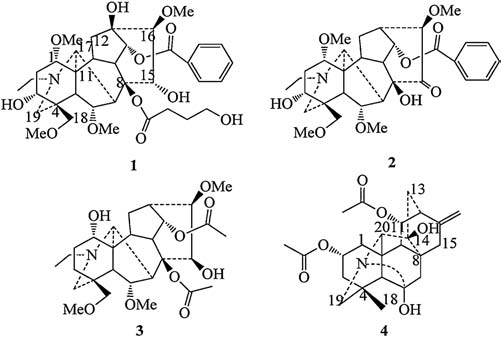
Nagaconitine A (1) was isolated as a colorless powder and showed positive reaction with the Dragendorff's reagent. The molecular formula of 1 was determined to be C36H51NO12 based on ESI-MS ([M + H]+; m/z 690) and HR-ESIMS (m/z 690.3484 [M + H]+, calcd. for 690.3478). IR spectrum showed the absorption for hydroxyl (3482, 3476 cm-1), conjugated ester carbonyl (1721 cm-1) and aromatic ring (1631, 1452 cm-1). The 1H and 13C NMR data displayed the presence of one N-ethyl group (δH 2.35, 2.86, m, each 1H; 1.07, t, 3H, J = 7.1 Hz; δC 46.9 t, 13.3 q), one benzoyl group (δH 8.02, d, 2H, J = 7.2 Hz; 7.57, dd, 1H, J = 7.2, 7.2 Hz; 7.45, dd, 2H, J = 7.2, 7.2 Hz; δC 165.9 s, 133.3 d, 129.7 d, 129.7 s, 128.7 d) and four OMe groups (δH 3.75, 3.28, 3.24, 3.15, each 3H, s; δC 61.3, 59.0, 58.2, 55.9 q×4). Compound 1 revealed 36 resonances in the 13C NMR (DEPT) spectrum (Table 1) attributing to 5 methyls, 8 methylenes, 16 methylidynes and 7 quaternary C-atoms, suggesting that compound 1 might be a C19- diterpenoid alkaloid [7, 8], bearing four OMe groups, one NCH2CH3, one OBz, and one OCO (CH2) 3OH (4-hydroxylbutyryl) unit. The 4- hydroxylbutyryl unit was confirmed by the cross-peaks between the signal at δH 1.56, 1.92 (each 1H, m, H-2') and C-1' (δC 175.0 s), C- 3' (δC 27.0 t), and C-40 (δC 61.5 t) in the HMBC spectrum (Fig. 2). The 1H and 13C NMR data (Tables 1 and 2) of compound 1 were almost the same as those of aconitine [9] except that the 4-hydroxylbutyryl unit signal appeared in compound 1 instead of an OAc group, suggesting that there was only one 4-hydroxylbutyryl group at C-8. The 2D NMR spectra including HMQC, HMBC, COSY and ROESY resulted in the assignments of all the protons and carbon atoms of compound 1 (Tables 1 and 2).

|
Download:
|
| Figure 2. Key 1H-1H-COSY (—) and HMBC (→) correlations of compounds 1-4. | |
|
|
Table 1 13C NMR data (100MHz, in CDCl3) of compounds 1-4. |
|
|
Table 2 1H NMR data (400 MHz, in CDCl3) of compounds 1-4. |

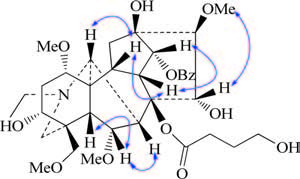
Compound 1 had the same relative configuration as aconitine, based on their almost same 1H NMR and 13C NMR data, and ROESY spectrum (Fig. 3). Thus, the structure of compound 1 was determined as 8- (4'-hydroxylbutyryl) aconitine, named Nagaconitine A (1).

|
Download:
|
| Figure 3. Key ROESY correlations of compound 1. | |
Nagaconitine B (2) had a molecular formula of C32H43NO9 in agreement with ESI-MS ([M + H]+ m/z 586) and HR-ESIMS (586.3011 [M + H]+, calcd. for 586.3007) analyses. IR bands exhibited hydroxyl (3441 cm-1), romatic ring (1632, 1452 cm-1) and carbonyl (1714 cm-1) functions. In the 1H NMR spectra, four OMesinglets (δH 3.76, 3.30, 3.25, 3.25, s, each 3H), an N-ethyl group (δH 2.40, m; 2.88, m; 1.05, t, J = 7.1 Hz, N-CH2CH3) and a benzoyl group (δH 7.97, d, J = 7.2 Hz; 7.61, dd, J = 7.2, 7.2 Hz; 7.45, dd, J = 7.2, 7.2 Hz) were observed, which exhibited characteristic features of an aconitine type C19-diterpenoid alkaloid. Comparing the 1H and 13C NMR spectral data (Tables 1 and 2) of compound 2 with those of 13-deoxyludaconitine [10] showed high similarity except that a C = O signal at δC (211.7 s) at C-15 appeared in 2, and the chemical shift due to C-15 was downfield shifted from δC 41.4 (t) to δC 211.7 (s), indicating that there was an C = O group located at C-15. This was futher confirmed by the long-rang correlations between H-16 (δH 3.84, and C-13 (δC 38.7 d), and C-15 (δC 211.7 s) in the HMBC spectrum. Accordingly, compound 2 was established to be 15-oxo-13-deoxyludaconitine.
Nagaconitine C (3) was elucidated as C28H43NO9 by ESI-MS ([M + H]+ m/z 538) and HR-ESIMS (538.3011 [M + H]+, calcd. for 538.3007) experiments. The 1H NMR and 13C NMR data displayed the signals of three OMe groups at δH 3.31, 3.31, 3.22 (each s, 3H), one N-ethyl signalat δH 1.13 (t, J = 7.2 Hz) and twoOAcgroupsat δH 2.13, 1.92 (each 3H, s). Careful analyses of the 1H and 13C NMR spectral data suggested that the structrue of compound 3 was similar to that of delphisine [11]. The main diference between the two compounds was that compound 3 contained one OH group at C-15. The long-range HMBC spectrum (Fig. 2) from H-15 (δH 5.14, 1H, d, J = 8.5 Hz) to C-8 (δC 74.2 s) and C-16 (δC 81.7 d) confirmed the location of the OH group. The correlations between H-16, assumed to be a-oriented with reference to the aconitine type C19- diterpenoid alkaloid [12], and H-15 were not observed in the ROESY spectrum, indicating the β-orientation of H-15. Hence, Nagaconitine C (3) was elucidated as 15-hydroxyldelphisine (3).
Nagaconitine D (4) was obtained as a colorless powder with a molecular formula C24H31NO6 deduced from its ESI-MS ([M + H]+ m/z 430) and HR-ESIMS (430.2224 [M + H]+, calcd. for 430.2221) analyses. The 1H NMR spectral of 4 exhibited three Me signals for a quaternary Me group (δH 1.41, s, H-18) and two OAc groups (δH 2.06, 2.03, s× 2). Its 13C NMR spectral data (Table 1) exhibited 24 C-atom signals, indicating that compound 4 might be a C20-diterpenoid alkaloid [13], bearing two OAc groups. The 1H and 13C NMR data (Tables 1 and 2) of compound 4 were almost identical with those of spiradine C [14] except that an OH group and an OAc signal appeared in compound 4. The OAc group was assigned to C-2 according to the HMBC spectrum in which the cross-peaks between H-2 (δH 5.22, br. s) and C-1 (δC 27.8 t), C-3 (δC 38.7 t), and C = O (δC 169.7 s) emerged. Moreover, the OH group locating at C-14 was also confirmed by the HMBC correlation between H-20 (δH 3.23 br. s) and H-13 (δH 2.00, 2.41, dd, J = 9.3, 5.4 Hz), and C-14 (δC 78.5 s) (Fig. 2). Consequently, Nagaconitine C (4) was defined as 14-hydroxyl-2-acetoxyspiradine C (4).
Compounds 1-4 were tested against human ovarian cancer cell line SK-OV-3 (Table 3). As a result, compounds 3-4 showed significant cytotoxicity against the tested cell line with inhibition of 31% (IC50 43.78 mmol/L) and 24% (IC50 32.14 μmol/L), respectively, as compared to the reference drug cisplatin (89%) with the IC50 values of 11.58 μmol/L.
|
|
Table 3 Cytotoxicity of compounds 1-4 against human ovarian cancer cell line SK-OV-3. |

{kind=link}
{kind=link}
{kind=link}
3. Conclusion
Four new diterpenoid alkaloids, nagaconitines A-D (1-4), were isolated from the roots of Aconitum nagarum var. heterotrichum. Their structures were elucidated by extensive spectroscopic examinations. Compounds 3-4 showed inhibition of cancer cell line SK-OV-3.
4. Experimental 4.1. General experimental proceduresOptical rotations were determined on a Jasco model 1020 polarimeter (Horiba, Tokyo, Japan). UV spectra were measured on a Shimadzu UV2401PC spectrophotometer (Shimadzu, Kyoto, Japan). IR (KBr discs, cm-1) spectra were recorded on a Bio-Rad FTS-135 spectrometer (Bio-Rad, Hercules, California, USA). 1D and 2D NMR were recorded on Bruker AM-400, Bruker DRX-500 or AVANCE III-600 spectrometers (Bruker, Bremerhaven, Germany). Mass spectra were run on a VG Spec-3000 spectrometer (VG, Manchester, UK) and Waters AutoSpec Premier P776 (Waters, USA). Silica gel (200-300 mesh) for column chromatography and TLC plates (GF254) were obtained from Qingdao Makall Chemical Company (Makall, Qingdao, China). Al2O3 for column chromatography was purchased from Shanghai Wusi Chemical Reagents Company, Ltd. (Wusi, Shanghai, China). Fractions were visualized by silica gel plates sprayed with dragendorff0s reagent.
4.2. Plant materialThe roots of Aconitum nagarum var. heterotrichum were collected in Weixi county of Yunnan Province, China, in September 2014, and authenticated by Prof. Qing-Er Yang from South China Botanical Garden, Chinese Academy of Sciences. A voucher specimen (No. YAU20140906) had been deposited in the College of Agriculture and Biotechnology, Yunnan Agricultural University.
4.3. Extraction and isolationThe air-dried roots of A. nagarum var. heterotrichum (11 kg) were powdered and extracted three times with MeOH for 2 h under reflux. The crude extract was dissolved with 8 L 2% HCl solution, then basified to pH 9.0 with ammonia (25%) and extracted with CHCl3 to obtain crude alkaloidal extract (256 g). The alkaloidal extract was performed on silica column chromatography (SiO2 CC, 1500 g, 12×100 cm), eluting with petroleum ether (30-60℃) -acetone-diethylamine (30:1:1, 20:1:1, 20:3:1, 20:6:1, 20:10:1, 10:20:1, v/v) to afford six fractions (A-F). Fr. C (43 g) was further separated to obtain five subfractions (C1-C5) by SiO2 CC with petroleum ether-acetone-diethylamine (20:1:1, each 500 mL). Fr. C2 (8.7 g) was performed on Al2O3 CC (2.0×35 cm, 30 g) and eluted with petroleum ether-EtOAc (6:1, each 100 mL) to yield compound 2 (25 mg). Fr. C3 (3.6 g) was applied to Al2O3 CC (4×40 cm, 140 g) with an eluent of petroleum ether-EtOAc (5:1, each 50 mL), and then purified through Sephadex LH - 20 (CHCl3/ MeOH, 1:1) to produce compounds 3 (63 mg) and 4 (37 mg). Fr. D (39 g) was further separated to obtain four subfractions (D1-D4) by Si CC with petroleum ether-acetone-diethylamine (15:3:1, v/v, each 500 mL) as the eluent. Fr. D2 (8.2 g) was applied to Al2O3 CC (5.0×45 cm, 320 g) with an eluent of petroleum ether-EtOAc (2:1, each 150 mL), and further purified through Sephadex LH-20 (CHCl3/MeOH, 1:1) to provide compound 1 (43 mg). All obtained compounds had a degree of purity > 90%, based on the TLC method in three different solvent systems exhibiting one spot with dragendorff0s reagent, and NMR spectra with the smooth baseline and no impurity peak.
Nagaconitine A (1) : Colorless powder; [α]D25 : + 2.00 (c 0.10, MeOH); UV (MeOH) : 230 (4.08) nm; IR (KBr, cm-1) vmax: 3482, 3476, 2931, 1721, 1631, 1452, 1098, 712. 1H NMR and 13C NMR data see Tables 1 and 2. HR-ESIMS at m/z 690.3484 [M + H]+ (calcd. for C36H52NO12, 690.3478).
Nagaconitine B (2) : Colorless powder; [α]D25 :-72.4 (c 0.10, MeOH); UV (MeOH) : 230 (4.10) nm; IR (KBr, cm-1) vmax: 3441, 2930, 1714, 1632, 1452, 1100, 714. 1H NMR and 13C NMR data see Tables 1 and 2. HR-ESIMS at m/z 586.3011 [M + H]+ (calcd. for C32H44NO9, 586.3007).
Nagaconitine C (3) : Colorless powder; [α]D25 : + 48.47 (c 0.10, MeOH); UV (MeOH) : 202 (3.28) nm; IR (KBr, cm-1) vmax: 3454, 2938, 1740, 1105. 1H NMR and 13C NMR data see Tables 1 and 2. HR-ESIMS at m/z 538.3011 [M + H]+ (calcd. for C28H44NO9, 538.3007).
Nagaconitine D (4) : Colorless powder; [α]D25 :-13.73 (c 0.10, MeOH); UV (MeOH) : 202 (3.73) nm; IR (KBr, cm-1) vmax: 3468, 3434, 2944, 1737, 1029. 1H NMR and 13C NMR data see Tables 1 and 2. HR-ESI-MS at m/z 430.2224 [M + H]+ (calcd. for C24H32NO6, 430.2221).
4.4. Antitumor assay in vitroCompounds 1-4 were tested for their acitvities against human ovarian cancer cell line SK-OV-3 by the MTT method [6]. Briefly, 100 mLofcellsuspension (5×104 cells/mL) wasseededinto96-well microtiter plates and cultured for 24 h before the compound was added. Then, differentconcentrations of compounds 1-4 were added to the plates, the cells were cultivated for 48 h, and 20 mL of MTT (5 mg/mL) was added to each well. After 4 h, the culture medium was removed and the formazan crystals were completely dissolved with 150 μL DMSO in each well by vigorously shaking the plate. Finally, the absorbance was measured at 570 nm on a Molecular Devices ELX-800 Microplate spectrophotometer (Omega Bio-Tek, Inc., Norcross, GA, USA). Cisplatin was used as a positive control.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (Nos. 21362045, 81560622) and the project of Sciences and Technology Department of Yunnan Province (No. 2012CG007). The authors are grateful to the staff of analytical group at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences for the spectral data.
| [1] | Q.E. Yang, Taxonomic notes on some species of Aconitum L. (Ranunculaceae) from Yunnan, China. Acta Phytotaxonom. Sin. 37 (1999) 545–590. |
| [2] | Yunnan Institute of Botany, Flora Yunnanica, Vol. 11, Science Press, Beijing., Science Press: Beijing, 1977p : 87 . |
| [3] | N.V. Mody, S.W. Pelletier, S.Y. Chen, The structure and partial synthesis of nagarine. A novel alkaloid from the Chinese drug, Aconitum nagarum var. heterotrichum F. Dielsianum W.T. Wang. Heterocycles. 17 (1982) 91–94. DOI:10.3987/S-1982-01-0091 |
| [4] | L.S. Ding, F.E. Wu, Y.Z. Chen, Diterpenoid alkaloids of Aconitum nagarum var. Heterotrichum. Nat. Prod. Res. Dev. 6 (1994) 50–54. |
| [5] | J.Y. Dong, Z.Y. Li, L. Li, Diterpenoid alkaloids from Aconitum nagarum var. lasiandrum. Chin. Chem. Lett. 11 (2000) 1005–1006. |
| [6] | Y.Q. Ge, R.B. Cheng, Y.H. Zhou, Cryptotanshinone induces cell cycle arrest and apoptosis of multidrug resistant human chronic myeloid leukemia cells by inhibiting the activity of eukaryotic initiation factor 4E. Mol. Cell. Biochem. 368 (2012) 17–25. DOI:10.1007/s11010-012-1338-3 |
| [7] | B.S. Joshi, S.K. Srivastava, A.D. Barber, H.K. Desai, S.W. Pelletier, Selective demethylation of some aconitine-type norditerpenoid alkaloids. J. Nat. Prod. 60 (1997) 439–443. DOI:10.1021/np9606862 |
| [8] | J.B. Hanuman, A. Katz, 1H-Nmr spectra of norditerpenoid alkaloids. A review, J. Nat. Prod. 57 (1994) 1473–1483. DOI:10.1021/np50113a001 |
| [9] | T. Mori, H. Bando, Y. Kanaiwa, K. Wada, T. Amiya, Studies on the constituents of Aconitum species. II. Structure of deoxyjesaconitine. Chem. Pharm. Bull. 31 (1983) 2884–2886. DOI:10.1248/cpb.31.2884 |
| [10] | C.S. Peng, F.P. Wang, X.X. Jian, New norditerpenoid alkaloids from Aconitum hemsleyanum var. pengzhouense. J. Asian Nat. Prod. Res. 2 (2000) 245–249. DOI:10.1080/10286020008041362 |
| [11] | S.W. Pelletier, Z. Djarmati, Carbon-13 nuclear magnetic resonance:aconitine-type diterpenoid alkaloids from Aconitum and Delphinium species. J. Am. Chem. Soc. 98 (1976) 2626–2636. DOI:10.1021/ja00425a036 |
| [12] | F.P. Wang, Q.H. Chen, The C19-diterpenoid alkaloids, in:G.A. Cordell (Ed.), The Alkaloids:Chemistry and Biology, Vol. 69, Elsevier, New York, 2010, pp. 1-577. |
| [13] | F.P. Wang, X.T. Liang, C20-diterpenoid alkaloids, in:G.A. Cordell (Ed.), The Alkaloids:Chemistry and Biology, Vol. 59, Elsevier, New York, 2002, pp. 1-288. |
| [14] | G. Goto, K. Sasaki, N. Sakabe, Y. Hirata, The alkaloids obtained from Spiraea japonica L.fil. Tetrahedron Lett. 9 (1968) 1369–1373. DOI:10.1016/S0040-4039(01)98955-5 |