 2017, Vol. 28
2017, Vol. 28 



b State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
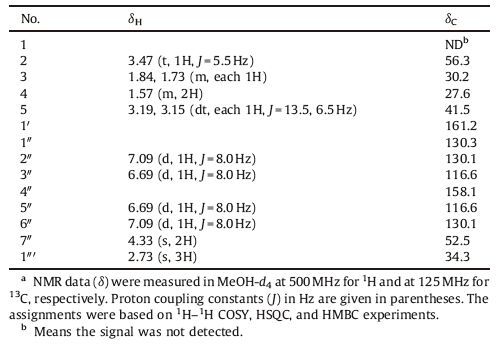
The genus Gymnadenia (Orchidaceae), containing about 20 species, is mainly distributed in Asia and Europe. Gymnadenia conopsea R. Br., native to the provinces of Tibet, Xinjiang, Inner Mongolia, Sichuan, Qinghai, and Gansu of mainland China, is a very important traditional Tibetan medicinal plant. The dried tuber of this plant, known as "Wangla" (Chinese), has been traditionally used for treatment of cough, asthma, and other syndromes [1-3]. Besides phenanthrenes, stilbenes, cyclodipeptides, and cyclopentene derivatives, a small group of glycosyloxybenzyl 2-isobutylmalate derivatives have been reported previously from this plant, which showed neuroprotective effects on memory deficits and pathological changes in senescent mice [3-6]. The current study was begun to search for novel bioactive secondary metabolites from this plant, and has led to the isolation of a new ureido-substituted amino acid, conopsamide A (1) (Fig. 1). This paper deals with the isolation, structure elucidation and HDAC1 (Histone Deacetylase 1) inhibitory activity of the new compound.

|
Download:
|
| Figure 1. Structure of 1. | |
2. Experimental 2.1. General experimental procedures
Optical rotations were measured on a P-2000 polarimeter. UV spectra were recorded on a V-650 spectrometer. IR spectra were recorded on a Nicolet 5700 FT-IR Microscope spectrometer (FT-IR Microscope Transmission). 1D NMR and 2D NMR spectra were obtained at 500 for 1H and 125 MHz for 13C, respectively, on an Inova 500 MHz spectrometer with solvent peaks as references. ESIMS and HR-ESIMS data were obtained on an AccuToFCS JMST100CS spectrometer. Column chromatography (CC) was performed with silica gel (200-300 mesh, Qingdao Marine Chemical Inc., China) and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden). HPLC separation was performed on a system consisting of a Lab Alliance prep pump and a Model 500 absorbance detector using an YMC (250 mm×10 mm i.d.) semi-preparative column packed with C18 silica gel (5 μm). TLC was conducted on precoated silica gel GF254 plates. Spots were visualized under UV light (254 or 356 nm) or by spraying with 10% H2SO4 in 95% EtOH followed by heating.
2.2. Plant materialThe tubers of G. conoposa were collected at Hezuo, Gansu Province, People's Republic of China, in September 2013. The plant identification was verified by Professor Lu Xuefeng, Northwest Institute of Plateau Biology, Chinese Academy of Sciences. A voucher specimen (No. 00769) is deposited at the Herbarium of College of Pharmaceutical Sciences, Qinghai University for Nationalities.
2.3. Extraction and isolationThe air-dried tubers of this plant (1.5 kg) were successively extracted with 5.0 L of 95% EtOH (48 h) and 5.0 L of 65% EtOH (2×48 h) at room temperature. The combined extracts were evaporated under reduced pressure to yield a residue (121 g). The residue was suspended in H2O (600 mL) and then successively partitioned with petroleum ether (3×500 mL) and EtOAc (3×500 mL). The aqueous phase was applied to a HP-20 macroporous adsorbent resin column (20 cm×200 cm), and eluted successively with H2O (3 L), 50% EtOH (10 L), and 95% EtOH (3 L) to yield three corresponding fractions A, B and C. After removing the solvent under reduced pressure, fraction B (0.3 kg) was separated by column chromatography (CC) over MCI gel CHP 20P (1 L), with successive elution using H2O (2 L), 30% EtOH (2 L), 50% EtOH (2 L), 95% EtOH (1 L), and Me2CO (1 L), to give fractions B1-B5. Fraction B2 (3.6 g) was fractioned by reversed-phase C-18 silica flash chromatography eluting with a step gradient from 20% to 80% MeOH in H2O, to afford fractions (B21-B26) based on TLC analysis. Separation of B23 (135mg) was separated by semipreparative HPLC (RP18, 5 μm, 222 nm, MeOH-H2O, 10:90) to give 1 (16.0 mg).
Conopsamide A (1): White amorphous; UV (MeOH) λmax (log ε)201 (3.68), 222 (3.79), 270 (2.21) nm; IR vmax 3332 (broad), 2928, 1694, 1668, 1591, 1537, 1513, 1447, 1392, 1233, 1167, 1102, 1012, 952, 821 cm-1; 1H NMR (MeOH-d4, 500 MHz) data, see Table 1; 13C NMR (MeOH-d4, 125 MHz) data, see Table 1; (+)-ESIMS m/z 318 [M + Na]+ and m/z 334 [M + K]+; (-) HR-ESIMS 294.1461 [M-H]-(calcd. for C14H20N3O4, 294.1459).
|
|
Table 1 NMR spectroscopic data for 1a. |
{kind=link}
2.4. Acid hydrolysis of 1 and Marfey's analysis of the hydrolyzates
Conopsamide A (1 mg) was hydrolyzed in 1.0 μL of 6 mol/L HCl at 120 ℃ for 12 h. Excess aqueous HCl was removed under vacuum. The dry material was suspended in 100 μL of H2O, and then treated with 1% FDAA in acetone (100 μL) and 1 mol/L NaHCO3 in 60 μL of water at 40 ℃ for 1 h. After cooling to room temperature, the mixture was neutralized with 2 mol/L HCl (30 μL) and filtrated. The standard FDAA-amino acids were prepared in the same manner using the corresponding L-and D-amino acids (0.8 and 1.1 mg). The FDAA-amino acid derivatives from the hydrolysate were compared with standard FDAA-amino acids using HPLC analysis: ZORBAX SB-C18 (4.6 mm×150 mm, 5 μm); flow rate, 1.0 mL/min; UV detection at 340 nm; column temperature, 20 ℃; mobile phase, a linear gradient of 9:1 50 μmol/L triethylammonium phosphate (TEAP) buffer (pH 3)/CH3CN to 1:9 TEAP/CH3CN over 90 min. The FDAA derivatives of amino acids liberated from 1 showed a peak at 31.013 min, matching the retention time of FDAA D-ornithine (31.0=min; 32.246 min for L-ornithine).
2.5. HDAC1 inhibition screen of 1HDAC1 (Cisbio Bioassays No. NP_0049=.2) inhibitory activity was measured using 2 ng/μL HDAC1, 0.2 μmol/L H3(1-21) K9 incubated for 60 min at 37 ℃ with a range of concentrations of compound 1. Reaction was stopped by addition 5 μL SA-XL665 and 2 ng H3K9me0 antibody. Then, fluorescence signal was measured on a fluorescence microplate reader with an emission wavelength of 665 nm. All dilutions were prepared in a buffer containing 50 mmol/L Tris-HCl, pH 8.0; 137 mmol/L NaCl; 2.7 mmol/L KCl; 1 mmol/L MgCl2; and 0.01% Tween 20.
3. Results and discussionCompound 1 was obtained as a white amorphous, [α]D20-38.7 (c 0.10, MeOH). The IR spectrum of 1 showed absorption bands for hydroxyl and amino (3332 cm-1), carbonyl (1694 and 1668 cm-1), and aromatic ring (1591 and 1513 cm-1) functionalities. The positive mode ESIMS of 1 exhibited a quasimolecular ion peak at m/z 318 [M + Na]+ and 334 [M + K]+, and the molecular formula C14H21N3O4 was determined by HR-ESIMS at m/z 294.1461 [M-H]-× (calcd. for C14H20N3O4, 294.1459), combined with the NMR data (Table 1). The 1H NMR spectrum of 1 in MeOH-d4 showed characteristic signals attributable to 4-substitued benzyl alcohol at δH 7.09 (d, 2H, J=8.0 Hz, H-2″, 6″), 6.69 (d, 2H, J=8.0 Hz, H-3″, 5″), and 4.33 (s, 2H, H2-7″) and an N-methyl singlet at δH 2.73 (3H, s, H3-1″′). In addition, it showed signals assignable to one nitrogenbearing methine at δH 3.47 (1H, t, J=5.5 Hz, H-2), one nitrogenbearing methylene at δH 3.19, 3.15 (dt, each 1H, J=13.5, 6.5 Hz, H-5a, 5b), and two methylene at δH 1.84, 1.73 (m, each 1H, H-3a, 3b), and 1.57 (m, 2H, H2-4). The 13C NMR spectrum displayed 13 carbon resonances corresponding to the above units and one carbonyl carbon at dC 161.6 (C-1′) (Table 1). The six degrees of unsaturation inherent in the molecular formula of 1, coupled with data showing the presence of one carbonyl group, indicated that 1 must possess another carbonyl carbon (C-1) instead of one ring, since one of the carbon signal was absent in the 13C NMR spectrum. Therefore, the above spectroscopic data, coupled with the consideration of chemical shifts of the proton and carbon resonances, indicate that 1 is an amino acid with a 4-hydroxybenzyl and N-methyl moieties, of which the structure was further elucidated by comprehensive analysis of 2D NMR data.
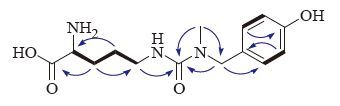
The proton and proton-bearing carbon resonances in the NMR spectra of 1 were unambiguously assigned by the HSQC experiment. In the 1H-1H COSY spectrum of 1, vicinal coupling correlations of H-2″′ (H-6″′)↔H-3″′ (H-5″′) (Fig. 2, thick lines) and HMBC correlation from H2-7″′ to C-2″′ (C-6″′) (Fig. 2, arrows), confirmed the presence of the 4-hydroxybenzyl. The 1H-1H COSY correlations of H-2↔H2-3 and H2-3↔H2-4, and H2-4↔H2-5 and the HMBC correlations from H2-3 to C-1 and C-5, from H2-4 to C-2 (Fig. 2, arrows), together with chemical shifts of the proton and carbon resonances, indicated the presence of an ornithinyl group. Finally, the connection of the 4-hydroxybenzyl, ornithinyl, and methyl via an ureido linkage was unambiguously established by HMBC correlations from H2-5 to C-1′, from N-methyl (H3-1″) to C-1′ and C-7″, and from H2-7″ to C-1″′ and C-1″′ (Fig. 2, arrows), completing the full planar structure of conopsamide A (1).

|
Download:
|
| Figure 2. Main 1H-1H gCOSY (thick lines) and HMBC correlations (blue arrows, from 1H to 13C) of 1. | |
{kind=link}
The absolute configurations of the a-amino residue in conopsamide A was determined by application of the Marfey's method [7]. Hydrolysis of conopsamide A (1) using 6 mol/L HCl yielded the free amino acid. The hydrolysis products were derivatized using the Marfey's reagent, (1-fluoro-2, 4-dinitrophenyl)-5-L-alanine amide (FDAA), and analyzed by reversed-phase HPLC. Peaks in the chromatogram were identified by comparing the retention times with those of the FDAA derivatives of authentic amino acids. Marfey's reagent derivative of the amino acid liberated from 1 showed peaks matching D-ornithine (Fig. S7 in Supporting information). Therefore, the structure of 1 was determined as (-)-R-2-amino-5-{[3-(4-hydroxybenzyl)-3-methyl] ureido}pentanoic acid, and named conopsamide A.
In the in vitro bioassays performed in this study, conopsamide A (1) showed moderate inhibitory activity against HDAC1 with an IC50 of 5.0 μmol/L. Other assays assessed inhibitory activity against the release of glucuronidase in rat polymorphonuclear leukocytes (PMN) induced by PAF [8]; cytotoxicity against several human cancer cell lines [9]; and inhibitory activity against protein tyrosine phosphatase 1B (PTP1B) [10]. However, conopsamide A (1) were inactive at a concentration of 10 × 10-6 mol/L in each assay.
4. ConclusionA new ureido-substituted amino acid, conopsamide A (1), was isolated from the ethanolic extract of G. conopsea tubers, and it showed moderate inhibitory activity against HDAC1. It is well known that over expression of HDCA promotes the deacetylation of histone in cancer cells, and enhances the attractiveness between DNA and histone by regaining the positive charges of histone. HDAC inhibition prevents the removal of acetyl groups from histones, causing their accumulation in cell nuclei and resulting in several downstream effects including apoptosis, differentiation, and reduced proliferation of cancer cells [11, 12]. The new structure with inhibitory activity against HDAC1 provides a framework for derivatives synthesis and further in deep biological evaluation.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (No. 81522050), Beijing outstanding talents cultivation fund (No. 2013D009008000002) and the Natural Science Foundation of Qinghai Province (No. 2014-ZJ-923) is acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.08.005.
| [1] | Beijing Institute of Botany, The Chinese Academy of Sciences, Iconographia Cormophytorum Sinicorum, Tomus V, vol. 5, Science Press, Beijing, 1976p. 631. |
| [2] | Jiangsu New Medical College, Dictionary of Traditional Chinese Medicine, vol. 1, ShanghaiScience andTechnology Publishing House, Shanghai, 1977, pp. 436-437. |
| [3] | J.C. Zi, S. Li, M.T. Liu, Glycosidic constituents of the tubers of Gymnadenia conopsea. J. Nat. Prod. 71 (2008) 799–805. DOI:10.1021/np070670j |
| [4] | H. Matsuda, T. Morikawa, H.H. Xie, M. Yoshikawa, Antiallergic phenanthrenes and stilbenes from the tubers of Gymnadenia conopsea. Planta Med. 70 (2004) 847–855. DOI:10.1055/s-2004-827234 |
| [5] | T. Morikawa, H.H. Xie, H. Matsuda, M. Yoshikawa, Glucosyloxybenzyl 2-Isobutylmalates from the tubers of Gymnadenia conopsea. J. Nat. Prod. 69 (2006) 881–886. DOI:10.1021/np0581115 |
| [6] | J.C. Zi, S. Lin, C.G. Zhu, Y.C. Yanga, J.G. Shi, Minor constituents from the tubers of Gymnadenia conopsea. J. Asian Nat. Prod. Res. 12 (2010) 477–484. DOI:10.1080/10286020.2010.491476 |
| [7] | P. Marfey, Determination of D-amino acids. II. Use of a bifunctional reagent, 5-difluoro-2, 4-dinititrobenzene. Carlsberg Res. Commun. 49 (1984) 591–596. DOI:10.1007/BF02908688 |
| [8] | X.N. Fan, J.C. Zi, C.G. Zhu, Chemical constituents of Heteroplexis micocephala. J. Nat. Prod. 72 (2009) 1184–1190. DOI:10.1021/np900213w |
| [9] | W.X. Song, S. Li, S.J. Wang, Pyridinium alkaloid-coupled secoiridoids from the flower buds of Lonicera japonica. J. Nat. Prod. 71 (2008) 922–925. DOI:10.1021/np800040k |
| [10] | S.Y. Mo, S.J. Wang, G.X. Zhou, Phelligridins C-F:cytotoxic pyrano[4, 3-c] [2] benzopyran-1, 6-dione and furo[3, 2-c]pyran-4-one derivatives from the fungus Phellinus igniarius. J. Nat. Prod. 67 (2004) 823–828. DOI:10.1021/np030505d |
| [11] | R.W. Johnstone, Histone-deacetylase inhibitors:novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1 (2002) 287–299. DOI:10.1038/nrd772 |
| [12] | O. Khan, N.B. La Thangue, HDAC inhibitors in cancer biology:emerging mechanisms and clinical applications. Immunol. Cell Biol. 90 (2012) 85–94. DOI:10.1038/icb.2011.100 |