 2017, Vol. 28
2017, Vol. 28 



b Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peaking Union Medical College, Beijing 100050, China
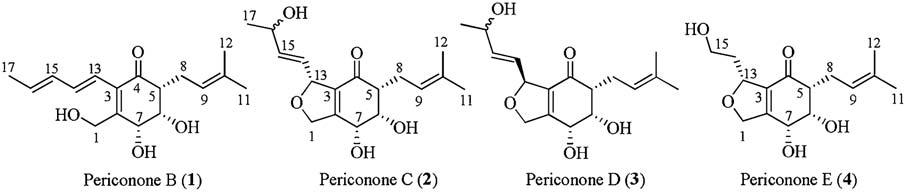
The term "meroterpenoid" is referred to as natural product of mixed biosynthetic origin that is partially derived from terpenoids [1]. Moreover, the structural diversity of meroterpenoids can be grouped into two major classes based on their biosynthetic origins: polyketide-terpenoids and non-polyketide-terpenoids. In recent years, a huge range of structurally diverse meroterpenoids were isolated from fungi [2] and displayed important biological activities, such as antimicrobial [3], immunomodulatory [4-6], antiviral [7], antitumoral [8-10], and antiinsect [11]. As part of our ongoing work of searching for novel secondary metabolites with interesting biological activities from the endophytic fungus Periconia sp. F-31 [12-16], a further chemical investigation led to the isolation of four meroterpenoids periconones B-E (1-4, Fig. 1). These compounds were proposed to be derived from typical polyketide-terpenoid hybrids pathway, which were formed from one acetyl-CoA starter and five malonyl-CoA extenders coupled with one C5 unit [17-20]. Herein, we report their isolation, structural elucidation, biological activities, and plausible biogenetic pathway.

|
Download:
|
| Figure 1. Structures of Periconones B-E (1-4). | |
2. Experimental 2.1. General experimental procedures
Optical rotations were measured on a PerkinElmer Model-343 digital polarimeter. The CD spectra were recorded on a JASCO J-815 spectropolarimeter. The UV absorption spectra were measured in MeOH on a Thermo Spectronic-Vision32 Software V1.25. IR spectra were acquired on a Nicolet 5700 FT-IR microscope spectrometer (FTIR Microscope Transmission). 1D and 2D NMR spectra were obtained at 600 MHz for 1H NMR and 150 MHz for 13C NMR on VNOVA SYSTEM-600 and Bruker AVIIID 600, and 400 MHz for 1H NMR and 100 MHz for 13C NMR on Bruker AVIII 400 spectrometers. Chemical shifts (δ) are given in ppm, and coupling constants (J) are given in hertz (Hz). HRESIMS data were measured using an Agilent Technologies 6520 Accurate Mass QTOF LC/MS spectrometer. Silica gel (200-300 mesh, Qingdao Haiyang Chemical Co. Ltd., Qingdao, China) and Sephadex LH-20 gel (Amersham Biosciences, Sweden) were used for column chromatography (CC). Semi-preparative reversed phase and normal phase HPLC were performed on a Shimadzu HPLC instrument equipped with a Shimadzu RID-10A detector and a Grace Adsorbosphere C18 column (250 mm × 10 mm, i.d., 5 μm) by eluting with mixtures of methanol and H2O at 3 mL/min, or a Grace Allsphere silica column (250 mm × 10 mm, i.d., 5 μm) by eluting with mixtures of n-hexane-isopropyl alcohol and n-hexane-EtOAC at 4 mL/min, respectively. Analytical TLC was carried out on pre coated silica gel GF254 plates (Qingdao Marine Chemical Industry, Qingdao, China), and spots were visualized under UV light or by spraying with 10% H2SO4 in EtOH followed by heating at 120 ℃.
2.2. Fungal material, fermentation, extraction and isolationThe fermentation, extraction, and isolation of the fungal strain Periconia sp. F-31 were performed as described previously [21]. The EtOAc extract (25.0 g) of the culture filtrate was subjected to silica gel CC eluting with a CH2Cl2-CH3OH gradient (100:0-0:100) to produce eight fractions (Fr1-Fr8) on the basis of TLC analysis. Fr3 (4.5 g) was initially subjected to Sephadex LH-20 CC by eluting with CH3OH to give four fractions (Fr3.1-Fr3.4). Fr3.4 (1.0 g) was then fractionated by reversed-phase semi-preparative HPLC eluting with CH3OH-H2O (65:35, v/v) to afford four fractions (Fr3.4.1-Fr3.4.4). Purification of Fr3.4.4 (78.9 mg) by reversed-phase semi preparative HPLC (CH3OH-H2O, 70:30, v/v) resulted in 1 (48.0 mg, tR 16.7 min). Fr4 (3.4 g) was fractionated by Sephadex LH-20 CC eluting with CH3OH, yielding five fractions (Fr4.1-Fr4.5). Fr4.4 (500 mg) was further separated via normal-phase semi-prepara tive HPLC (n-hexane-EtOAC, 2:3, v/v) to furnish four fractions (Fr4.4.1-Fr4.4.4). Purification of Fr4.4.1 (120.0 mg) through re versed-phase semi-preparative HPLC (CH3OH-H2O, 30:70, v/v) followed by normal-phase semi-preparative HPLC afforded 2 (4.2 mg, tR 16.2 min, n-hexane-isopropyl alcohol, 4:1, v/v), 3 (1.5 mg, tR 28.5 min, n-hexane-isopropyl alcohol, 5:1, v/v), and 4 (2.2 mg, tR 15.2 min, n-hexane-isopropyl alcohol, 4:1, v/v).
Periconone B (1). Yellow gum; [α]54620 +21.0 (c 0.10, MeOH); UV (MeOH) λmax (logε) 206 (3.99) nm; CD (MeOH) λmax (Δε) 209 (-2.93), 254 (+1.64), 348 (-0.14) nm; IR (vmax) 3410, 2971, 2913, 1677 cm-1; 1H NMR and 13C NMR data, see Table 1; HR-ESIMS m/z 293.1741 [M+H]+ (C17H25O4, calcd. [M+H]+ 293.1753).
|
|
Table 1 1H NMR and 13C NMR data of periconones B-E (1-4). |
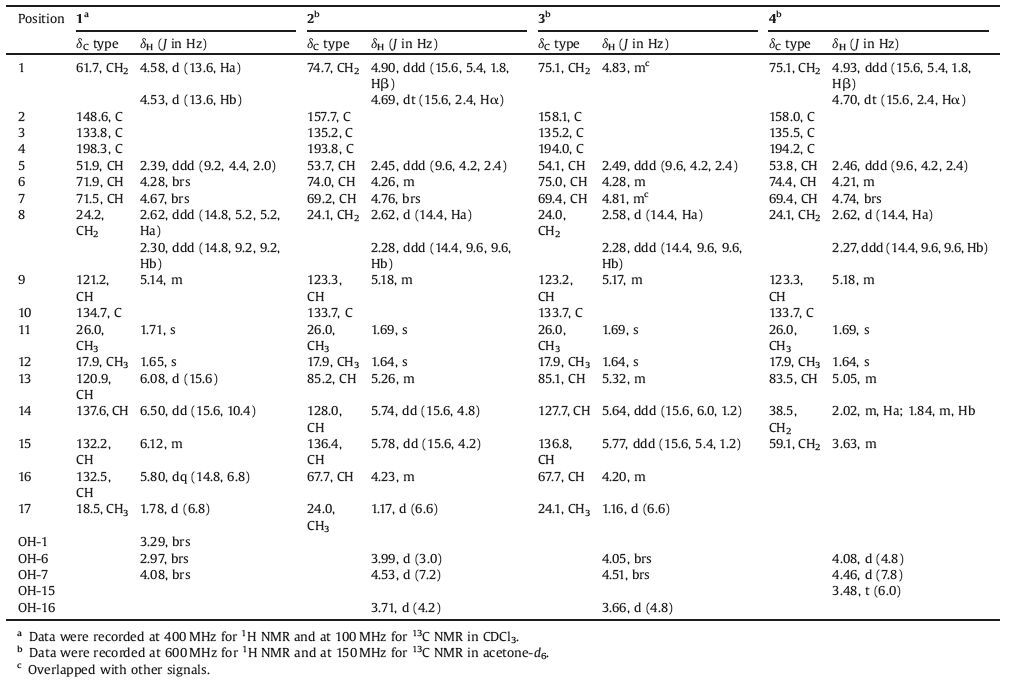
{kind=link}
Periconone C (2). Yellow gum; [α]54620 +40.0 (c 0.05, MeOH); UV (MeOH) λmax (logε) 204 (4.12) nm; CD (MeOH) λmax (Δε) 207 (-4.45), 245 (+4.81), 335 (-0.34) nm; IR (vmax) 3400, 2970, 2926, 1676 cm-1; 1H NMR and 13C NMR data, see Table 1; HR-ESIMS m/z 331.1506 [M+Na]+ (C17H24O5Na, calcd. [M+Na]+ 331.1516).
Periconone D (3). Yellow gum; [α]54620 +73.0 (c 0.10, MeOH); UV (MeOH) λmax (logε) 204 (4.04), 243 (3.65) nm; CD (MeOH) λmax (Δε) 256 (+3.42), 328 (-0.54) nm; IR (vmax) 3422, 2974, 2918, 1675 cm-1; 1H NMR and 13C NMR data, see Table 1; HR-ESIMS m/z 331.1505 [M+Na]+ (C17H24O5Na, calcd. [M+Na]+ 331.1516).
Periconone E (4). Yellow gum; [α]54620 +21.4 (c 0.07, MeOH); UV (MeOH) λmax (logε) 248 (3.74) nm; CD (MeOH) λmax (Δε) 204 (-6.92), 2=(+5.14), 311 (-0.10) nm; IR (vmax) 3404, 2921, 1673 cm-1; 1H NMR and 13C NMR data, see Table 1; HR-ESIMS m/z 283.1533 [M+H]+ (C15H23O5, calcd. [M+H]+ 283.1540).
2.3. Cytotoxicity bioassay [22]The cytotoxicity of the compounds against the human tumor cell lines (HCT-8, Bel-7402, Hela, and MCF-7) was measured using the MTT assay. The cells were maintained in a RRMI S7 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/mL streptomycin. Cultures were incubated at 37 ℃ in a humidified atmosphere of 5% CO2. Tumor cells were seeded in 96-well microtiter plates at 1200 cells/well. After 24 h, compounds were added to the wells. After incubation for 96 h, cell viability was determined by measuring the metabolic conversion of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) into purple formazan crystals by viable cells. The MTT assay results were read using an MK3 Wellscan (Labsystem Dragon, Helsinki, Finland) plate reader at 570 nm. All compounds were tested at five concentrations (10-5 mol/L, 10-6 mol/L, 10-7 mol/L, 10-8 mol/L, and 10-9 mol/L) in 100% DMSO with a final concentration of DMSO of 0.1% (v/v) in each well. Paclitaxel was used as a positive control. Each concentration of the compounds was tested in three parallels experiments. IC50 values were calculated using Microsoft Excel software.
2.4. HIV-inhibitory bioassay [23]293Tcells (2 ×105) were co-transfected with 0.6 μg of pNL-LuvE--Vpu- and 0.4 μg of pHIT/G. After 48 h, the VSV-G pseudotyped viral supernatant (HIV-1) was harvested by filtration through a 0.45 μm filter and the concentration of viral capsid protein was determined by p24 antigen capture ELISA (Biomerieux). SupT1 cells were exposed to VSV-G pseudo typed HIV-1 (MOI=1) at 37 ℃ for 48 h in the absence or presence of test compounds (Efavirenz was used as positive control). The inhibition rate was determined by using a firefly Luciferase Assay System (Promega).
3. Results and discussionPericonone B (1) was obtained as a yellow gum and gave an HRESIMS ion peak at m/z 293.1741 [M+H]+, corresponding to a molecular formula of C17H24O4 with six degrees of unsaturation. The IR absorption bands at 3410 cm-1 and 1677 cm-1 indicated the presence of hydroxyl and conjugated carbonyl groups. The 1H NMR spectrum of 1 (Table 1) in CDCl3 showed five olefinic protons at δH 6.50 (dd, 1H, J=15.6, 10.4 Hz), 6.12 (1H, m), 6.08 (d, 1H, J=15.6 Hz), 5.80 (dq, 1H, J=14.8, 6.8 Hz), and 5.14 (1H, m); one oxygenated methylene protons at δH 4.58 (d, 1H, J=13.6 Hz) and 4.53 (d, 1H, J=13.6 Hz); two oxygenated methine protons at δH 4.67 (1H, brs) and 4.28 (1H, brs); one methylene protons at δH 2.62 (ddd, 1H, J=14.8, 5.2, 5.2 Hz) and 2.30 (ddd, 1H, J=14.8, 9.2, 9.2 Hz); one methine proton at δH 2.39 (ddd, 1H, J=9.2, 4.4, 2.0 Hz); three methyl groups at δH 1.78 (d, 3H, J=6.8 Hz), 1.71 (3H, s), and 1.65 (3H, s), and three hydroxyl groups at δH 2.97 (brs, 1H), 3.29 (brs, 1H), and 4.08 (brs, 1H). The 13C NMR and DEPT spectra (Table 1) exhibited 17 carbon resonances, including four quaternary carbons (one α, β-unsaturated ketone at δC 198.3, three olefinic), eight methines (including five olefinic, two oxygenated), two methylenes (including one oxygenated), and three methyls. Among the 17 carbons, one carbonyl carbon and four double bonds accounted for five degrees of unsaturation, indicating that compound 1 possessed one ring system.
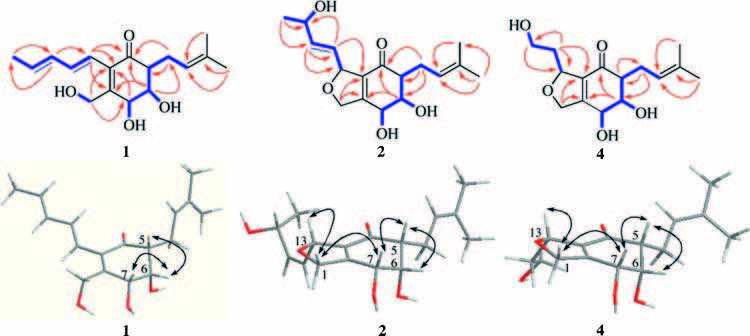
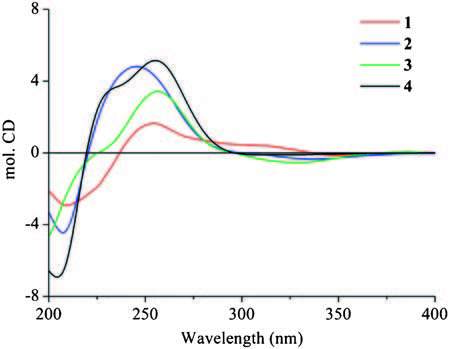
The HMBC correlations (Fig. 2) from H-5 to C-4, C-6, and C-7; from H-6 to C-2, C-4, C-5, and C-7; from H-7 to C-2, C-3, and C-6; and from H-1 to C-2, C-3, and C-7, together with experiment four spin systems C (5) H-C (6) H-C (7) H; OH-6/H-6; OH-7/H-7; and OH-1/H2-1 shown in the 1H-1H COSY, established a cyclohexenone ring unit, which was substituted with a hydroxymethyl group at C-2 and two hydroxyl groups at C-6 and C-7, respectively. In addition, four olefinic proton resonances at δH 5.80 (H-16), 6.12 (H-15), 6.50 (H-14), and 6.08 (H-13) with the large coupling constants of JH-15, H-16=14.8 Hz and JH-13, H-14=15.6 Hz, indicated the presence of two E-olefins, and the consecutive 1H-1H COSY correlations from H-13 to H-17 revealed the presence of a 1, 3-pentadienyl side chain. The HMBC correlations of H-13/C-2, C-3, and C-4 supported the location of the 1, 3-pentadienyl side chain at C-3 of cyclohexenone. Furthermore, the 1H-1H COSY and HMBC relationships established a prenyl unit, which was linked to C-5 according to the HMBC interactions of H-8 (δH 2.62 and 2.30) with C-4, C-5, and C-6 (Fig. 2). In NOESY experiments, the enhancement of H-5 and H-7 when H-6 was irradiated, along with the small coupling constants of JH-5, H-6 and JH-6, H-7, suggested the syn-orientation of H-5, H-6, and H-7. The CD spectrum of 1 (Fig. 3) showed the characteristic Cotton effect (CE) of the α, β-unsaturated ketone, the configuration at C-5 was determined as R based on a negative CE at 348 nm (the n-π* transition of the α, β-unsaturated ketone) [24, 25]. On the basis of this relative configuration, the absolute configuration at C-6 and C-7 were assigned as S and R, respectively. Thus, the structure of compound 1 was established.

|
Download:
|
Figure 2. 1H-1H COSY   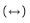 |
|
{kind=link}

|
Download:
|
| Figure 3. The experimental ECD spectra of Periconones B-E (1-4). | |
{kind=link}
Periconone C (2) was obtained as a yellow gum. Its molecular formula was established as C17H24O5 according to the HR-ESIMS peak at m/z 331.1505 [M+Na]+ (calcd. for C17H24O5Na, 331.1516) with six degrees of unsaturation. The IR spectrum indicated the presence of hydroxyl (3400 cm-1) and conjugated carbonyl (1676 cm-1) groups. Detailed comparison of the NMR data of 2 with those of 1 revealed the similarity of the resonance signals in cyclohexenone and prenyl unit, but significant differences in the side chain of C-3, in which two olefinic carbon signals disappeared and two additional oxygen-bearing carbons at δC 85.2 and 67.7 were observed in 2. The H3-17 (δH 1.17, d, J=6.6 Hz), H-15 (δH 5.78, dd, J=15.6, 4.2 Hz), and H-14 (δH 5.74, dd, J=15.6, 4.8 Hz) displayed the HMBC cross peaks with C-16 (δC 67.7) with a connected methine proton at δH 4.23 (H-16, m) on the basis of HSQC experiment. These results indicated the attachment of a hydroxyl group at C-16 and the presence of one E-olefin at the side chain of C-3, which were further confirmed by the 1H-1H COSY correlation of OH-16 (δH 3.71, d, J=4.2 Hz)/H-16 and the HMBC correlations of H-14/C-13, C-15, C-16, and C-3; H-15/C-13, C-14, and C-16. Furthermore, the chemical shifts of C-13 (δC 85.2) and C-1 (δC 74.7) together with the molecular formula indicated the attachment of an ether linkage between C-1 and C-13. In NOESY experiments, the enhancement of H-7 and H-6 when H-5 was irradiated, together with the small coupling constants of JH-5, H-6 and JH-6, H-7 suggested the syn-orientation of H-5, H-6, and H-7. The NOESY correlations of Hβ-1/H-7 and Hβ-1/H-13 supported that H-7 and H-13 were b-orientation. Interestingly, the absolute configuration of C-16 noted to have no effect on the CD spectrum (Fig. 3) of the cyclohexenone ring in 2, the remaining chiral carbons were assigned as 5R, 6S, 7R, 13R by the characteristic CE of the α, β-unsaturated ketone (a negative CE at 335 nm) [24, 25]. Owing to the limited amount of 2 available for the determination of configuration at C-16, the stereo-configuration of C-16 still remains to be determined.
Periconone D (3) was obtained as a yellow gum and had the same molecular formula as 2 as determined by the HR-ESIMS peak at m/z 331.1505 [M+Na]+ (calcd. for C17H24O5Na, 331.1516). The general features of its 1H NMR and 13C NMR spectroscopic data closely resembled those of 2 except that the signals for H2-1, H-13, and C-1 revealed a chemical shift difference compared with those of 2 (Table 1). Analysis of DEPT, 1H-1H COSY and HMBC correlations suggested that 3 was a stereoisomer of 2. In NOESY experiments, the enhancement of H-6 and H-7 when H-5 was irradiated suggested the syn-orientation of H-5, H-6, and H-7. However, due to the chemical shifts of Hα-1 and Hβ-1 in close proximity and overlapped with H-7 in 1H NMR spectrum with acetone-d6 as solvent, the relative configuration of H-13 was not assigned. Fortunately, the proton signals were separated each other when the NMR solvent was switched to CDCl3. The NOESY correlations of Hβ-1/H-7 and Hα-1/H-13 indicated the opp-orientation of H-13 and H-7. The negative CE at 328 nm was observed for 3 in the CD spectrum (Fig. 3 and Fig. S35 in Supporting information), which was similar to those of 1 and 2, indicating 5R configuration for 3. The appearance of obvious different CE at 207 nm should be attributable to the π-π* transition of the E-olefin at the side chain of C-13 [26], which further suggested the presence of stereoisomer at C-13 in compounds 2 and 3. Accordingly, the absolute configuration of 3 was determined as 5R, 6S, 7R, 13S. The absolute configuration of C-16 still remains to be resolved.
Periconone E (4) was obtained as a yellow gum with a molecular formula of C15H22O5 determined from the HR-ESIMS peak at m/z 283.1533 [M+H]+ (calcd for C15H23O5, 283.1540). The IR spectrum showed the presence of hydroxyl (3404 cm-1) and conjugated carbonyl (1673 cm-1) groups. The general features of its 1H NMR and 13C NMR spectroscopic data (Table 1) were similar to those of 2 except for the side chain of C-13. The disappearance of the signals of doublet methyl (H3-17) and oxygenated methine (H-16) were observed, and the double bond between C-14 and C-15 was replaced by those of sp3 C-14 (δH 2.02 and 1.84; δC 38.5) and C-15 (δH 3.63; δC 59.1) in compound 4. Moreover, the 1H-1H COSYcorrelation of OH-15 (δH 3.48, t, J=6.0 Hz)/H-15, suggested the presence of C-15 alcohol, which was also consistent with its HRESIMS data. The structure of 4 was further confirmed by DEPT, 1H-1H COSY, HSQC, and HMBC experiments (Fig. 1). In NOESY experiments, the enhancement of H-6 and H-7 when H-5 was irradiated, together with the small coupling constants of JH-5, H-6 and JH-6, H-7 suggested the syn-orientation of H-5, H-6, and H-7. The NOESY correlations of H-13/Hβ-1, and H-7/Hβ-1 indicated the β-orientation of H-7 and H-13. The absolute configuration of C-5 in 4 was determined to be R on the basis of n-π* transition of the α, β-unsaturated ketone (negative CE at 311 nm, Fig. 3) [24, 25], on the basis of the relative configuration, the remaining chiral carbons, C-6, C-7, and C-13 were assigned as 6S, 7R, 13R.
All these compounds were evaluated for in vitro cytotoxicity against human tumor cell lines (HCT-8, Bel-7402, HelA, and MCF-7) by using an MTT method and anti-HIV activities. Compound 4 exhibited cytotoxicity against the MCF-7 tumor cell line with an IC50 value of 4.2 μmol/L (paclitaxel as the positive control, IC50=0.2 nmol/L). Compound 1 displayed anti-HIV activity with an IC50 value of 18.0 μmol/L, while efavirenz (positive control) gave an IC50 value of 1.4 nmol/L.
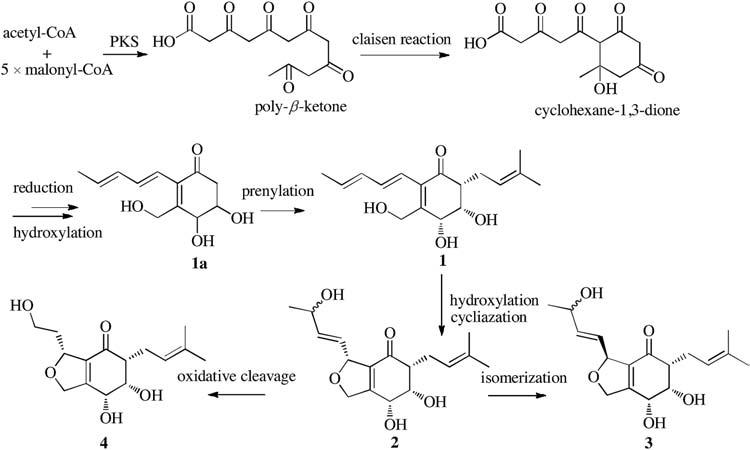
Periconones B-E (1-4) can be classified as polyketide-terpenoid hybrid molecules based on their proposed biosynthetic origins. A plausible biogenetic pathway suggested that the Periconones B-E shared the common polyketone intermediate (1a) originating from one acetyl-CoA starter and five malonyl-CoA extenders [17, 18]. Then 1a was prenylated to be 1 [19, 20], and followed by a selective subsequent modification such as cycliazation, hydroxylation, isomerization, and oxidative cleavage, compounds 2-4 were formed with different structure diversity (Scheme 1).

|
Download:
|
| Scheme 1. Proposed biosynthesis of periconones B-E (1-4). | |
{kind=link}
4. Conclusion
In this communication, we described the isolation and identification of four new merterpenoids periconones B-E (1-4) from the endophytic fungus Periconia sp. F-31. Biological studies showed that compound 1 exhibited anti-HIV activity with an IC50 value of 18.0 μmol/L and 4 displayed cytotoxic activity against the human MCF-7 tumor cell line with an IC50 value of 4.2 μmol/L.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.07.031.
| [1] | J.W. Cornforth, Terpenoid biosynthesis. Chem. Br. 4 (1968) 102–106. |
| [2] | R. Geris, T.J. Simpson, Meroterpenoids produced by fungi. Nat. Prod. Rep. 26 (2009) 1063–1094. DOI:10.1039/b820413f |
| [3] | T. Fujioka, K. Yao, K. Hamano, Epi-cochlioquinone A, a novel acyl-CoA:cholesterol acyltransferase inhibitor produced by Stachybotrys bisbyi. J. Antibiot. 49 (1996) 409–413. DOI:10.7164/antibiotics.49.409 |
| [4] | S. Omura, F. Kuno, K. Otoguro, Arisugacin, a novel and selective inhibitor of acetylcholinesterase from Penicillium sp. IFO-4259. J. Antibiot. 48 (1995) 745–746. DOI:10.7164/antibiotics.48.745 |
| [5] | W.M. Hu, S. Ohyama, K. Hasumi, Activation of fibrinolysis by SMTP-7 and -8, novel staplabin analogs with a pseudosymmetric structure. J. Antibiot. 53 (2000) 241–247. DOI:10.7164/antibiotics.53.241 |
| [6] | W.G. Kim, K.M. Cho, C.K. Lee, I.D. Yoo, Terreulactones A, B, C, and D:novel acetylcholinesterase inhibitors produced by Aspergillus terreus. II. Physicochemical properties and structure determination. J. Antibiot. 56 (2003) 351–357. DOI:10.7164/antibiotics.56.351 |
| [7] | K. Minagawa, S. Kouzuki, H. Tani, Novel stachyflin derivatives from Stachybotrys sp. RF-7260. Fermentation isolation, structure elucidation and biological activities. J. Antibiot. 55 (2002) 239–248. DOI:10.7164/antibiotics.55.239 |
| [8] | X.L. Yang, C. Qin, F. Wang, Z.J. Dong, J.K. Liu, A new meroterpenoid pigment from the basidiomycete Albatrellus confluens. Chem. Biodivers. 5 (2008) 484–489. DOI:10.1002/(ISSN)1612-1880 |
| [9] | D.B. Stierle, A.A. Stierle, J.D. Hobbs, J. Stokken, J. Clardy, Berkeleydione and berkeleytrione, new bioactive metabolites from an acid mine organism. Org. Lett. 6 (2004) 1049–1052. DOI:10.1021/ol049852k |
| [10] | D.B. Stierle, A.A. Stierle, B. Patacini, The berkeleyacetals, three meroterpenes from a deep water acid mine waste Penicillium. J. Nat. Prod. 70 (2007) 1820–1823. DOI:10.1021/np070329z |
| [11] | R. Geris, E. Rodrigues-Fo, H.H.G. Silva, I.G. da Silva, Larvicidal effects of fungal meroterpenoids in the control of Aedes aegypti L., the main vector of dengue and yellow fever. Chem. Biodivers. 5 (2008) 341–345. DOI:10.1002/(ISSN)1612-1880 |
| [12] | H.L. Ge, D.W. Zhang, L. Li, Two new terpenoids from endophytic fungus Periconia sp. F-31. Chem. Pharm. Bull. 59 (2011) 1541–1544. DOI:10.1248/cpb.59.1541 |
| [13] | D.W. Zhang, X.Y. Tao, J.M. Liu, A new polyketide synthase-nonribosomal peptide synthetase hybrid metabolite from plant endophytic fungus Periconia sp. Chin. Chem. Lett. 27 (2016) 640–642. DOI:10.1016/j.cclet.2016.02.005 |
| [14] | D.W. Zhang, X.Y. Tao, J.M. Liu, Periconiasin G, a new cytochalasan with unprecedented 7/6/5 tricyclic ring system from the endophytic fungus Periconia sp. Tetrahedron Lett. 57 (2016) 796–799. DOI:10.1016/j.tetlet.2016.01.030 |
| [15] | D.W. Zhang, X.Y. Tao, R.D. Chen, Pericoannosin A, a polyketide synthasenonribosomal peptide synthetase hybrid metabolite with newcarbon skeleton from the endophytic fungus Periconia sp. Org. Lett. 17 (2015) 4304–4307. DOI:10.1021/acs.orglett.5b02123 |
| [16] | D.W. Zhang, H.L. Ge, J.H. Zou, Periconianone A, a new 6/6/6 carbocyclic sesquiterpenoid from endophytic fungus Periconia sp. with neural antiinflammatory activity. Org. Lett. 16 (2014) 1410–1413. DOI:10.1021/ol500197x |
| [17] | R.J. Cox, Polyketides, proteins and genes in fungi:programmed nano-machines begin to reveal their secrets. Org. Biomol. Chem. 5 (2007) 2010–2026. DOI:10.1039/b704420h |
| [18] | J. Staunton, K.J. Weissman, Polyketide biosynthesis:a millennium review. Nat. Prod. Rep. 18 (2001) 380–416. DOI:10.1039/a909079g |
| [19] | Y.H. Chooi, J.X. Fang, H. Liu, Genome mining of a prenylated and immunosuppressive polyketide from pathogenic fungi. Org. Lett. 15 (2013) 780–783. DOI:10.1021/ol303435y |
| [20] | M.E. Tanner, Mechanistic studies on the indole prenyltransferases. Nat. Prod. Rep. 32 (2015) 88–101. DOI:10.1039/C4NP00099D |
| [21] | D.W. Zhang, H.L. Ge, D. Xie, Periconiasins A-C, new cytotoxic cytochalasans with an unprecedented 9/6/5 tricyclic ring system from endophytic fungus Periconia sp. Org. Lett. 15 (2013) 1674–1677. DOI:10.1021/ol400458n |
| [22] | J. Carmichael, W.G. DeGraff, A.F. Gazdar, J.D. Minna, J.B. Mitchell, Evaluation of a tetrazolium-based semiautomated colorimetric assay:assessment of chemosensitivity testing. Cancer Res. 47 (1987) 936–942. |
| [23] | Q. Zhang, Z.L. Liu, Z.Y. Mi, High-throughput assay to identify inhibitors of Vpu-mediated down-regulation of cell surface BST-2. Antivir. Res. 91 (2011) 321–329. DOI:10.1016/j.antiviral.2011.07.007 |
| [24] | G. Snatzke, Circulardichroismus-IX:modifizierung der octantenregel für α, β-ungesättigte ketone:transoide enone. Tetrahedron 21 (1965) 421–438. DOI:10.1016/S0040-4020(01)98283-3 |
| [25] | D.N. Kirk, The chiroptical properties of carbonyl compounds, the chiroptical properties of carbonyl compounds. Tetrahedron 42 (1986) 777–818. DOI:10.1016/S0040-4020(01)87486-X |
| [26] | A.I. Scott, A.D. Wrixon, A symmetry rule for chiral olefins. Tetrahedron 26 (1971) 3695–3715. |