 2017, Vol. 28
2017, Vol. 28 



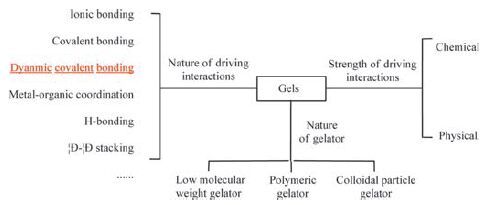
A gel is "a soft, solid or solid-like material consisting of two or more components one of which is a liquid, present in substantial quantity" according to Kramer’s definition [1-3]. Gels consist of three-dimensional cross-linked networks, typically surrounded by a large number of solvent molecules. They maintain shape like solid materials. Solvent is the major component, and the gelator is the minor component. Gels combine the elastic behaviour of solids with the microviscous properties of fluids. Gels are generally classified as chemical and physical gels according to the strength of the driving interactions involved in their formation (Fig. 1) . Chemical gels are usually tough and stable, whereas they are brittle, poorly transparent and unable to self-heal. In contrast physical gels are weak and show stimuli-responsibility because non-covalent interactions are susceptible to the external environment [4-6].

|
Download:
|
| Figure 1. Classification of gels by nature of driving interactions, strength of driving interaction and nature of gelator. | |
{kind=link}
Gelators can be considered as ‘functional’ molecules, polymeric precursors or colloidal particles in solution (Fig. 1) . The gelators aggregate via weak interactions to form one-dimensional (1D), 2D and 3D structures. For discrete molecules, 1D structures may be formed via weak interactions. For bridging molecular precursors, oligomeric/polymeric aggregates may be formed. Reversible reactions occur between these nanoscale aggregates. The solubility is important for gelation. If an insoluble polymer/aggregate is formed, amorphous precipitation or crystallization will occur in solution. The precipitation or crystallization can be considered as irreversible sequestration. For example, metal-organic frameworks (MOFs) and covalent-organic frameworks (COFs) are 2D and 3D polymeric materials that crystallize from the metal-organic and dynamic covalent systems, respectively. If the initially formed aggregates have a proper solubility in solution, there are two routes for further growing. First, the aggregates grow in one direction to form fibres. Second, the nanoparticles are interconnected via interactions to form a sponge-like porous structure. Either route can lead to gelation.
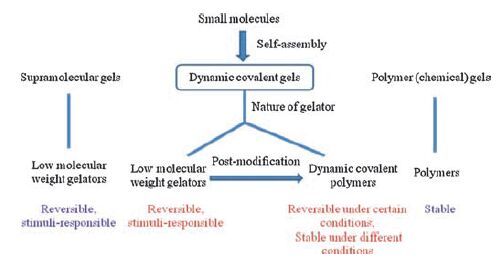
Physical gels of low molecular weight gelators (LMWGs) are commonly referred as supramolecular gels. Sometimes, supramolecular gels also include the gels that are induced by non-covalently cross-linked polymeric compounds (Fig. 1) . In supramolecular gels, the gelators are self-assembled via non-covalent interactions [7, 8]. Self-assembly of the gelators are typically under thermodynamic control, so it is possible to make intricate structures that are difficult to achieve by traditional stepwise synthesis. The interactions between the gelators are reversible, so supramolecular gels show responses to external stimuli. Supramolecular gels usually stay at room temperature and liquefy at high temperatures due to breaking of the intermolecular interactions [9-16].
In gels, the driving forces (a broad realm of interatomic forces) range from strong chemical bonds to weak intermolecular interactions (Fig. 1) . Strong chemical bonds like most covalent bonds are built irreversibly. The resulting chemical gels are robust, tolerant to physical deformation and thermally irreversible. It is difficult to change their structures after preparation. This catalogue of gels is dominated by kinetically controlled chemistry, e.g., covalently crosslinked gels obtained by radical polymerization. Various intermolecular non-covalent interactions are also driving forces for aggregation, such as electrostatic interactions (including hydrogen bonding [17-20], halogen bonding [21]), π-π interactions [22-25], van der Waals/hydrophobic forces [26-28] and host-guest interactions [29-31], multiple combined interactions and others [32-34]. Non-covalent interactions are commonly reversible and sensitive to environment. These gels are typically physical gels under thermodynamic control. Their states are readily changed by external stimuli (e.g., temperature, substance).
A number of dynamic bonds are receiving growing intention recently. Their strengths lie between strong chemical bonds and weak intermolecular interactions. They selectively undergo reversible breaking and reformation under equilibrium conditions. They are relatively weak in contrast to covalent bonds, but multiple non-covalent bonds work in concert to profoundly affect the final structure. Multiple non-covalent bonds usually exist between two or more components with functional units via hydrogen bonding, metal-ligand coordination or donor-acceptor interactions [35-41]. Dynamic covalent bonds are one typical category of dynamic bonds [42-48]. Dynamic covalent chemistry, which was introduced by Lehn, focuses on reversible reactions of making and breaking of covalent bonds under mild conditions [49]. Different from traditional covalent chemistry that has predominantly focused on the use of kinetically controlled reactions, dynamic covalent chemistry highlights thermodynamic assemblies and products. The relative proportions of the individual assemblies and products are decided by the differences in free energy between the transition states. Energetic differences are small among alternative structures, and product distributions depend on the relative stabilities of the final products. The reversibility introduces "proofreading" and "error checking" and allows the exchange of molecular components at equilibrium. It will assure that kinetically trapped imperfections are corrected and ultimately favour formation of the most stable structures.
Dynamic covalent bonds usually have higher bond strengths and more controllable reversibility than other non-covalent interactions. They have generally slower kinetics of bond cleavage and formation. The slow kinetics of most dynamic reactions typically results in microcrystalline powders and no efficient errorcorrection is allowed for the growth of single crystals in COFs [9, 10, 50-55]. Dynamic covalent bonds reversibly form and break under certain conditions (e.g., in the presence of catalyst), while they can be strong and permanent under different conditions. Dynamic covalent bonds can be further transformed to kinetically fix the dynamic exchange. For example, the C=N bonds in imines and acylhydrazones are reduced to form non-reversible covalent C-N bonds. So far some dynamic covalent reactions are able to be performed at ambient temperature with efficient error-proof construction under thermodynamic control, ability to self-repair and adaptability (response to external stimuli) like non-covalent interactions. Meanwhile these reactions have advantages over non-covalent interactions. Compared with metal-organic coordination, dynamic covalent bonds are more directional interactions. Dynamic covalent structures are more stable in aqueous environments than hydrogen-bonded structures. These features of dynamic covalent bonds allow for a great degree of control over the gelation process and thus greatly broaden the scope of gels.
A number of review articles have been devoted to supramolecular gels [9, 10, 50-55] and polymer gels [56-58], however, a comprehensive analysis of dynamic covalent bonding in gels is not available. The main purpose of this review is to discuss the role of dynamic covalent bonding in gels assembled from small molecules. Dynamic covalent reactions used in gels are reviewed first in order to understand the dynamic covalent bonding. The survey of the literature is classified into two catalogues according to the nature of gelators and the interactions between gelators: gelation by discrete molecules and gelation by dynamic covalent polymers. We aim to illustrate the structure-property relationships of various dynamic covalent gels and the link between primary interactions and bulk material properties. We hope to shed some light on the future work and inspire continuous endeavors in this area.
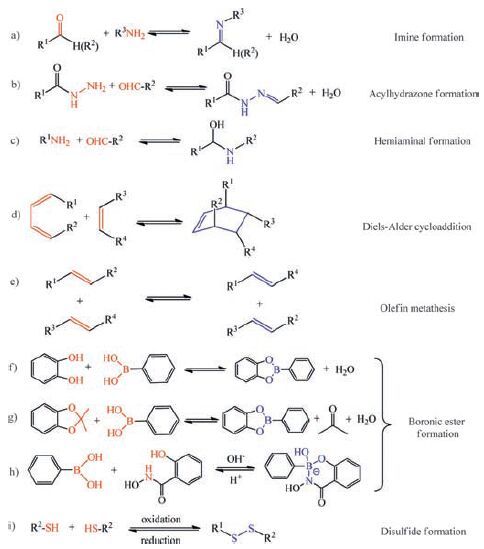
2. Dynamic covalent reactionsFor a dynamic covalent reaction, rapid equilibration is critical to reaching the thermodynamic minima in a reasonable time frame, and mild reaction conditions allow broader substrate scope [59]. The dynamic reactions that have been applied for gels include formation and exchange of imines, hydrazones, acylhydrazones, Diels-Alder cycloaddition, olefin metathesis, boronic esters, disulfides and so on (Scheme 1) . In some of dynamic covalent reactions (e.g., formation of imines and boronic esters), ancillary molecules (e.g., water) are formed.

|
Download:
|
| Scheme1. Part of reversible dynamic covalent reactions. | |
{kind=link}
One of the most widely used dynamic covalent reactions is condensations of amino and carbonyl groups to give C=N compounds (imines, hydrazones and oximes). Directionality of the C=N bonds can control the dynamic process. Among the C=N compounds, formation of imine bonds is one of the most frequently used motifs in dynamic covalent chemistry [60-64]. The imine bond is formed by the reversible condensation between a primary amine and an aldehyde or ketone (Reaction of aldehydes with amines can also reversibly form hemiaminals or aminals). The imine reaction is also called Schiff base condensation reaction. The imine reaction is acid (Lewis or Bro¨nsted acid) catalyzed. Imine formation generally takes place fast at pH 4-5 and slow at very low or very high pH. Imine bond has good stability under neutral or basic conditions. Imines are good ligands for metal ions. Imines with ortho-hydroxy substituents in the aromatic ring [65] and pyridine-containing imines [66, 67] have been widely studied for metal complexation.
Hydrazines and acylhydrazines form C=N derivatives of aldehydes and ketones, called hydrazones and acylhydrazones, respectively. Hydrazone C=N bonds are dynamic under mild acidic conditions or nucleophilic catalysts [68] but are stable (kinetically inert) under neutral and basic conditions.
Diels-Alder cycloaddition reaction and its retro-Diels-Alder analogue have been used in polymer materials (gels) [69-74]. Diels-Alder reaction enables the formation of two carboncarbon bonds between a conjugated diene (a 4π-electron system) and a dienophile compound containing a double bond (a 2pelectron system) to form a cyclic (bicyclic) adduct. Use of dienes and dienophile that have complementary electron-releasing and electron-donating properties enhance the rate of Diels-Alder reactions. Diels-Alder reaction is thermoreversible. At lower temperatures, the forward Diels-Alder reaction is favoured, resulting in C-C bond formation. At elevated temperatures, the retro-Diels-Alder reaction is favoured and the C-C bond is broken.
Olefin metathesis is catalyzed by a Ru-or Mo-based complex [75, 76]. Through a metallocyclobutane intermediate, the carboncarbon double bonds rapidly exchange under mild conditions. Olefin metathesis reactions are versatile, highly efficient and tolerate functional groups with appropriate catalysts [77-81].
Formation of boronic ester has also been used in the formation of gels. Boronic acid undergoes reversible trimerization or condensation reactions with 1, 2-or 1, 3-diols (catechol) to form boroxines or five-or six-membered cyclic boronic esters. Formation of boronic ester is more favourable at high pH or/and in the presence of electron poor aryl substituent on the boronic acid [82, 83].
Disulfide bonds are usually formed from the oxidation of thiols [84]. The disulfide bond is stable under neutral and acidic conditions, but disulfides exchange rapidly under reductive or basic conditions in the presence of thiolates. A variety of oxidants promote the disulfide formation including air, and disulfide exchange can be triggered by a catalytic amount of reductant such as dithiothreitol. In addition, disulfide metathesis is achieved by UV irradiation [85].
Besides the above reactions, reversible radical exchange reactions are widely used in dynamic covalent polymers [86]. Photomediated radical addition and fragmentation reaction allow chain rearrangement of cross-linked polymer network [87].
To enable reversibility, some reactions have to be carried out under harsher reaction conditions. For example, transesterification reactions turn into reversible exchange reaction in the presence of zinc acetyl acetonate as catalyst at high temperatures (e.g., 200 ℃) [88]. These harsh reactions conditions are not advantageous to tolerate the presence of other functionalities. Thus ideal dynamic covalent reactions should be reversible under relatively mild conditions.
For a dynamic covalent reaction the position of the equilibrium is sensitive to a number of external factors such as pH, temperature, solvent, concentration and others. Consequently the system incorporating dynamic covalent bonds shows adaptability and stimuli responsiveness. Heating usually accelerates the bond breaking and reforming rate. It displaces the equilibrium toward depolymerization for polymers, thus bringing fluidity and processability. The systems are sensitive to solvents. The chemical equilibrium is displaced toward network depolymerization and dissolution in the presence of a solvent. The equilibrium also depends on the initial concentration of precursors. However, kinetically trapped species may exist simultaneously, including insoluble intermediates and discrete species. These species undergo more rapid intramolecular than intermolecular interconversion due to high effective molarity [59, 89].
Dynamic covalent bonds respond to a variety of environmental changes. Le Châtelier’s principle applies in these equilibrium reactions. For example, removal of the starting material causes the product to revert back to reactant. Dynamic covalent bonds can be stimulated by temperature, light, electric fields, mechanical deformation, etc [90]. For example, benzoic imine bond is pHresponsive, and can be hydrolyzed under mildly acidic conditions. The imine bond can be kinetically fixed by quenching the acid catalyst or reducing the double bond. In fact the majority of dynamic covalent reactions require a suitable catalyst to facilitate reversible bond formation in order to reach the thermodynamic equilibrium. Thus the processes can be readily halted and the products can be fixed kinetically by quenching the catalyst. It is known dynamic covalent bonds are stable enough to maintain their structure in the absence of external stimuli, as in the case of conventional covalent polymers.
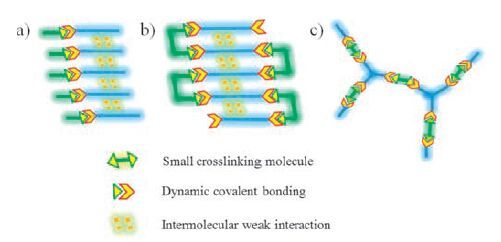
3. Design strategies for dynamic covalent gelsGels may be designed according to the nature of gelators and the interactions between gelators. In this review, design strategies for the gels that are formed from low molecular weight molecules are discussed. Low molecular weight molecules with functional groups may react via dynamic covalent bonding to form discrete molecular gelators or dynamic covalent polymer gelators. The gels are classified into two catalogues, gelation by discrete molecules and gelation by dynamic covalent polymers (Figs. 2 and 3) .

|
Download:
|
| Figure 2. Schematic representation of (a) dynamic covalent bonding incorporated in low molecular weight gelators, (b) post-modification of low molecular weight gels with crosslinking small molecules, (c) gelation by dynamic covalent polymers from functional small molecules. | |
{kind=link}

|
Download:
|
| Figure 3. Features of dynamic covalent gels and their comparison with supramolecular gels and polymer gels. | |
{kind=link}
Dynamic covalent bonding can be involved to form low molecular weight gelators. Discrete dynamic covalent gelators self-assemble to form gels through multiple non-covalent bonds (Fig. 2a). In these gelators, dynamic covalent bonding does not play a key role in the formation of gel matrices in some sense, which is better understood to mainly form discrete gelator compounds. Design of such gelators usually follows the known rules of organogelators [9, 10, 50-55]. The resulting discrete molecules aggregate via strong and directional intermolecular interactions to form thermoreversible physical gels. The gelation is achieved through hydrogen bonding, π-π stacking, and other supramolecular weak interactions. Such gels are readily reverted to the fluid state by applying external stimuli (e.g., heating) to break these supramolecular weak interactions. In the mean time, the introduction of dynamic covalent bonding in these gels allows further tuning of their stimuli-responsive properties. For example, heating affects the equilibrium position of dynamic covalent reactions, thereby also impacting on the structure and properties of dynamic covalent gels.
Dynamic covalent bonding is considered to be the primary force in the formation of 3D gel networks as low molecular weight molecules with multiple functional groups react to form dynamic covalent polymers that act as gelators (Fig. 2c). In these gels, low molecular weight molecules themselves are not gelators. Such covalent dynamic gels usually have infinitely extended structures, and thus cannot be redissolved upon heating and do not show thermoreversible gel-sol transitions. At the same time, another strategy is known to form dynamic covalent polymer gels via postmodification of low molecular weight gels. In the strategy, discrete low molecular weight gelators first self-assemble to form gels and subsequent crosslinking of discrete gelators via dynamic covalent bonding yields dynamic covalent polymers (Fig. 2b).
Dynamic covalent bonding is also involved in polymer gels [91]. In these gels, dynamic covalent bonding is employed to crosslink individual polymer chains to form 3D gel networks. The polymers can be crosslinked via dynamic covalent bonding between functional groups of the polymer chains; The polymer chains can also be crosslinked by small molecules. However, this catalogue of polymer gels is beyond the scope of this review.
4. Gelation by discrete moleculesMolecular gels are assembled from low molecular weight gelators. LMWGs assemble into entangled three-dimensional networks through weak intermolecular forces such as hydrogen bonding, π-π stacking, and van der Waals interactions. Such gels can be readily transformed into fluids by external stimuli (heating, sonicating etc.). Two classes of gels have been investigated according to the role of molecular precursors. One class requires all the added components to access the gel (i.e., no component forms the gel on its own). For the other class, at least one component can form the gel and other component can impact the assembly process and the gel’s properties [35]. For the first class of gels, gel phases can be generated by simply combining two structurally simple components. The gelator can be made in situ from soluble aqueous precursors via dynamic covalent bonds or synthesized beforehand. Incorporation of dynamic covalent bonding into molecular gels allows for fast screening of the gelation ability of a range of molecules under controlled and mild conditions.
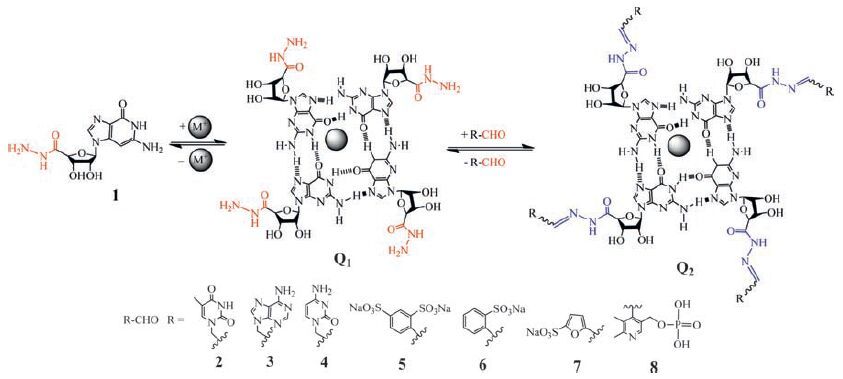
Based on formation of reversible hydrazone bonding, a class of guanosine hydrazide-based supramolecular hydrogels have been developed by Lehn and coworkers (Fig. 4) [92]. Guanosine hydrazide 1 is a powerful hydrogelator. It forms G-quartets Q1 in the presence of template cations such as Na+, K+, and NH4 +, which further self-assemble to form fibres and gels. The G-quartets Q1 are capable of undergoing dynamic decoration through formation of reversible acylhydrazone bonds with various aldehydes. In a range of aldehydes 2-8, the reactions of 1 with 1-formyl furan-3-sulfonic acid 7 and with pyridoxal-5-phosphate 8 yield gels of the acylhydrazone quartet derivatives Q2. In contrast, the aldehydes 2-4 disrupt the gel formed by 1 and result in product precipitation, and the aldehydes 5 and 6 give solutions. The gelation process results from a multilevel self-assembly based on the cation-templated self-assembly of quartets of guanosine acylhydrazone derivatives. This represents an example of gelationdriven self-organization with component selection and amplification in dynamic hydrogels based on G-quartet formation and reversible covalent connections.

|
Download:
|
| Figure 4. Reversible decoration of G-quartet assembly Q1 of guanosine hydrazide 1 via hydrazone formation with various aldehydes (2-8) . | |
{kind=link}
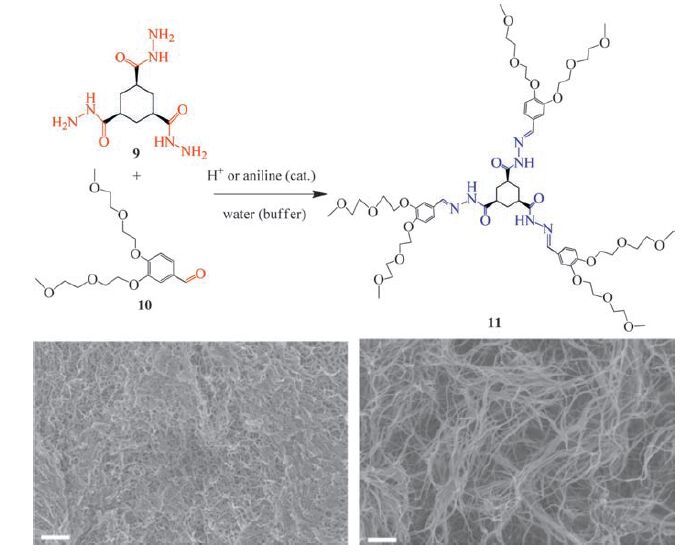
Reversible hydrazone bonding affect rheological characteristics and other properties of dynamic covalent gels. A number of gel materials with different mechanical strength can be produced starting from one initial solution mixture [93, 94]. For example, Van Esch, Eelkema and coworkers showed that an acylhydrazone gelator 11 is formed in situ by the reaction of the cyclohexane trishydrazide building block 9 and three molecules of aldehyde 10 (Fig. 5) [94]. Compound 11 self-assembles into elongated fibrous structures and forms an entangled gel network which is driven by hydrogen bonding and hydrophobic interactions. Acid or aniline catalysis enables rapid formation of 11 and its hydrogels under ambient conditions. This is an interesting example that catalytic action on the dynamic covalent bond can control the rate of assembly formation [95]. Remarkably, materials properties can be modulated by catalysis arising from the dynamic nature of acylhydrazone bonding. Using either acid or nucleophilic aniline catalysis drastic enhancement of mechanical strength of the hydrogels was observed. Under catalytic conditions a dense, heavily branched fibre network was obtained, whereas unbranched fibre bundles without catalyst. A higher rate of gelator formation leads to increased defects in the gel fibre, which results in branching of fibres and strengthens the resulting gel. A remarkable increase of the storage modulus (G’) was observed for the catalysed samples (uncatalyzed at pH 7.0, G’ = 5 kPa; catalysed, pH 5.0, G’ = 50 kPa). In addition, the gelation behavior can be controlled and fluorescent materials can be obtained by varying the chemical structure of the individually non-gelating starting components [96]. Remarkably a simple hydrazone molecule 12 (Scheme 2) is luminescent in the gel state due to the formation of two intramolecular H-bonds [97].

|
Download:
|
| Scheme2. Chemical structure of gelator 12. | |
{kind=link}

|
Download:
|
| Figure 5. Catalytic control of the formation of acylhydrazone gelator 11, and SEM images showing the hydrogel with aniline catalyst loading at pH 5.0 (left) and the hydrogel without catalyst at pH 7.0 (right) (scale bars 500 nm). Reproduced with permission from [94]. Copyright - 2013 Macmillan Publishers Limited. | |
{kind=link}
Imine bonding has also been used to construct discretemolecules [98] and involved in low molecular weight gelators. Cholesterol is one of the most versatile units utilized to design functional gelators and the corresponding ALS gelators are comprised of an aromatic (A) functional group coupled with a steroidal (S) moiety through a flexible linker (L). Cholesterol-appended aromatic imine organogelators 15a-15d are obtained from cholesterol-appended aniline 13 and 4-substituted aldehydes 14a-14d (Fig. 6) [99]. The resulting imine gelators from gels in various alcohols (n-butanol, n-pentanol, n-octanol). In contrast neither cholesterol-appended aniline 13 nor benzaldehydes 14a-14d gelate any solvents. SEM and AFM reveal the formation of long fibres on the order of millimeters. Selfassembly of the cholesterol-imine gelators are driven by the cooperative cholesterol-cholesterol and aromatic-aromatic stacking interactions. Additionally the fibres exhibit helicity arsing from the chiral feature of the cholesterol moiety.

|
Download:
|
| Figure 6. Formation of cholesterol-appended aromatic imine gelators, and AFM height images of the (a) 13 + 14a, (b) 13 + 14b, and (c) 13 + 14c gels and (d) AFM phase image of the 13 + 14c gel in n-BuOH (The bars are 0.5 mm each). Adapted with permission from [99]. Copyright - 2009 American Chemical Society. | |
{kind=link}
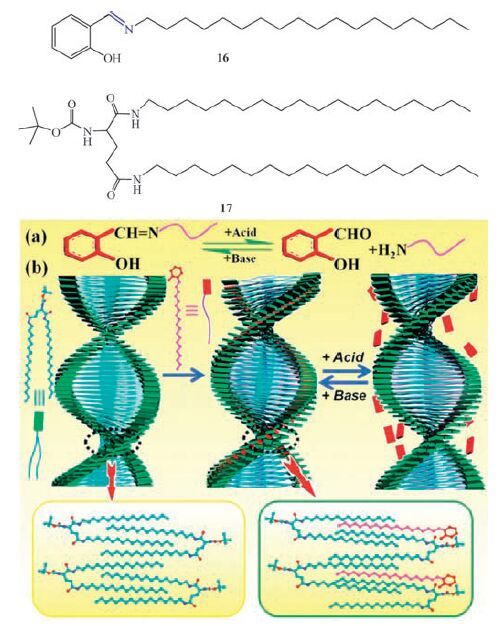
Chiroptical switch has been achieved through the formation and cleavage of the covalent imine bond [100]. An achiral imine amphiphile 2-((octadecylimino)methyl)phenol (16) does not form organogels in any kind of organic solvents, however 16 forms coorganogels when mixed with a universal gelator N, N’-bis(octadecyl)-L-bocglutamic diamide (17) in EtOH (Fig. 7) . Compound 17 self-assembles through hydrophobic interactions and H-bonding, and 16 is inserted into the alkyl chains of 17 molecules. Supramolecular chirality presumably comes from the chirality transfer from 17 through interchain interactions. It reveals that a chiral supramolecular organogel can be obtained from an achiral imine and a chiral gelator. Interestingly the chirality transfer was stopped once the imine bond was destroyed under acidic conditions. The chirality reappeared after the imine bond was subsequently recovered under alkaline conditions (by addition of Et3N).

|
Download:
|
| Figure 7. Schematic representation of formation of the chiral gel from achiral imine 16 and chiral gelator 17. | |
{kind=link}
The properties of imine gels can be readily controlled by metal ions [101-103] considering imines are good ligands for metal ions. 2-hydroxybenzylideneamino-N, N'-bisoctadecyl-lglutamicdiamide (18) and 4-hydroxybenzylideneamino-N, N'-bisoctadecyl-l-glutamicdiamide (19) are two compounds combining imine and amphiphilic glutamate with hydroxyl substituents at different positions (Fig. 8) [101]. Compound 18 has ability of intramolecular hydrogen-bonding interaction. Compound 18 forms organogels in many organic solvents owing to strong hydrogen-bonding interactions, π-π interactions and hydrophobic interactions. In contrast, for 19 the OH group at the para-position of the aromatic ring weakens its H-bonding ability and inhibits the π-π stacking of the aromatic rings, thus leading to no gelation. When divalent ions are introduced into the system the interaction modes are changed. Cu2+ ions and 18 react to form planar structure 18-Cu, which assembles into a helical twist. Cu2+ ions coordinate to the OH group of 19, and induce gelation. Mg2+ ions and 18 form a different complex 18-Mg. The 18-Mg gel shows chiral recognition towards D-tartaric acid. More favourable binding of D-tartaric acid on the gel nanofibres causes more significant quenching of the fluorescence of the 18-Mg gel than L-tartaric acid.

|
Download:
|
| Figure 8. Imine compounds 18, 19, 18-Cu and 18-Mg and their gelation behaviours. | |
{kind=link}
In another example of incorporating imine bonding into molecular gels, metal-assisted gelation was investigated by Bunzen and coworkers [102]. Imine metallogelators with cholesterol groups 21a-21c are in situ synthesised from steroidal amine 20, 2-pyridinecarboxaldehyde and metal ions (Cu2+, Ni2+, Zn2+) in a 3:3:1 ratio (Scheme 3) . The selected metal ions (Cu2+, Ni2+, Zn2+) are hexacoordinate with octahedral coordination geometry, thus the resulting gelator molecules contain three cholesterol groups. The building blocks spontaneously self-assemble around template metal ions leading to a simultaneous dynamic covalent (imine C=N) and coordination (N-metal) bond formation. Compounds 21a-21c induce gels in various alcohols (n-pentanol, n-hexanol, nheptanol, n-octanol) at room temperature. The gels are multiresponsive. Besides the common thermal stimulus, the gels are also sensitive to stoichiometry and chemical stimuli.

|
Download:
|
| Scheme3. Formation of imine metallogelators 21a-21c with cholesterol groups. | |
{kind=link}
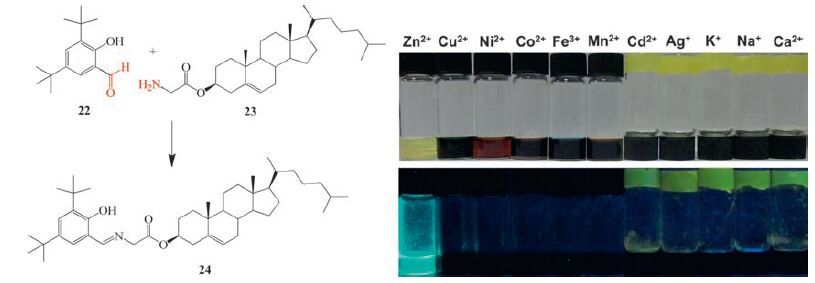
In the above examples imines show good coordination ability to metal ions and the properties of the gels are tuneable. Based on imine chemistry, a fluorescent gel sensor has been developed for metal ions [103]. An ALS-type imine gelator 24 is synthesized beforehand from cholesterol 2-aminoacetate (23) and 3, 5-di-tertbutyl-2-hydroxybenzaldehyde (22) (Fig. 9) . Compound 24 efficiently gelate various organic solvents such as alcohols, toluene, THF, EtOAc and 1, 4-dioxane. Nanofibres with the width ranging from 30 to 100 nm are responsible for the gelation. The gel shows thermochromism due to the enol-keto tautomerism. Interestingly the gel has aggregation-induced emission (AIE) with strong yellow green fluorescence, which is ascribed to a combination of inhibition of the intramolecular rotation and the formation of Jaggregates. In a range of metal ions, the gel shows selective dualresponsive property to Zn2+ through a gel-sol transition and a change in fluorescent turn-on. Additionally the gel also shows a two-channel response to F-by sol-gel transition and color changes because 24 is a strong H-bonding acceptor.

|
Download:
|
| Figure 9. Color changes (up) and fluorescent changes (bottom, under 365 nm light) of 24 gel (15 mg mL-1, in EtOH) to various metal ions (5 equiv.). | |
{kind=link}
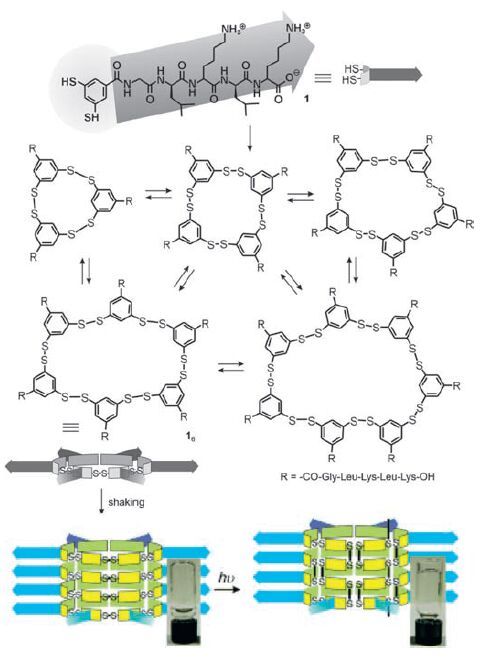
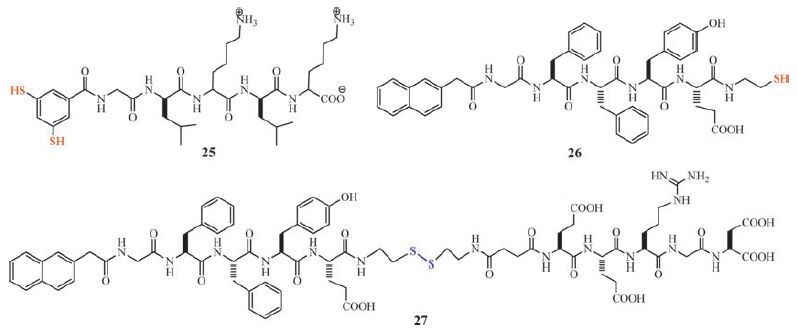
Disulfide dynamic chemistry has been involved in transition of peptide-derived hydrogels [104-107]. Otto and coworkers reported that a dithiol 25 equipped with a short peptide forms hexameric macrocycles upon oxidizing and agitation by shaking (Scheme 4, Fig. 10) [104]. The hexameric macrocycles selfassemble to form fibres as a free flowing aqueous solution. Subsequent photoirradiation of the solution can induce hemolytic cleavage of disulfides and result in disulfide exchange and formation of a hydrogel. The gelation is achieved by conversion of the hexameric macrocycles into polymers. This shows that the self-assembled structures are stabilized in a process akin to covalent capture [105-107]. It is worth mentioning that the actual gelator in this gel is dynamic covalent polymeric species (see below). In another example, Yang and coworkers reported that the cleavage of the disulfide bond in 27 (Scheme 4) leads to the formation of peptide hydrogel [108]. The hydrogelation of 26 is achieved by addition of reductants to PBS buffer solutions (pH 7.4) of 27. Using a similar strategy, they also developed Fmoc-short peptide-based supramolecular hydrogels for 3D cell culture of 3T3 cells [109].

|
Download:
|
| Figure 10. Self-assembly of peptide dithiol 25 and photo-induced gelation. | |
{kind=link}

|
Download:
|
| Scheme4. Chemical structures of peptide gelator 26 and precursors of peptide gelators 25 and 27. | |
{kind=link}
Interestingly the amide bond can exchange at ambient conditions and function as a dynamic covalent bond with a right enzyme catalyst [110, 111]. Reversing the amide bond formation normally requires extreme conditions (e.g., highly basic or acidic solutions and/or high temperature). However, Ulijn and coworkers reported that enzyme-assisted self-assembly of aromatic short peptide derivatives is fully reversible and equilibrium driven. A non-specific endoprotease (thermolysin) catalyzes peptide bond formation and hydrolysis between N-(fluorenyl-9-methoxycarbonyl) (Fmoc)-protected amino acids 28 (glycine, G; leucine, L; phenylalanine, F; threonine, T) with a fourfold excess of nucleophile 29 (G2, F2, L2 dipeptides or L-, F-OMe amino-acid esters) (Scheme 5) . This system initially appears as a milky suspension and forms a transparent self-supporting hydrogel over 60 min. During the process enzymes convert non-assembling precursors into selfassembling components, and nanoscale fibres are responsible for the gelation. A molecular model of the resulting structures shows that the Fmoc groups are stacked via π-π stacking and the peptide side chains arrange into an antiparallel β-sheet structure. The rearrangement of fluorenyl groups into extensive J-aggregates was evidenced by fluorescence spectroscopy and an increase in chiral ordering of fluorenyl groups was shown by circular dichroism. Through formation of the self-assembled structure, the amide component is relatively stabilized.

|
Download:
|
| Scheme5. Reversed hydrolysis reaction in which peptide derivatives are formed from an Fmoc-amino acid 28 and a dipeptide or amino acid ester 29. | |
{kind=link}
5. Gelation by dynamic covalent polymers
There are two methods to obtain dynamic covalent polymer gels from small molecules, post-modification of molecular gels (Fig. 2b) and direct gelation by dynamic covalent polymers (Fig. 2c). The methods allow formation of dynamic covalent polymers from relatively simple precursors. Dynamic covalent polymers exhibit dynamic properties such as self-healing, shape memory and environmental adaptation. Compared to noncovalent interactions, dynamic bonds are more robust with generally slower kinetics of bond cleavage and formation. Arising from their mechanically robust nature, this class of dynamic covalent gels can be developed as catalyst. The bottom-up approach for gel synthesis provides opportunity for the design of porous materials with various functionalities. After catalytically active moieties are immobilized in the gel matrix, dynamic covalent gels can be applied in supported catalysis [112, 113].
A preformed molecular gel may be post-modified via dynamic covalent crosslinking to obtain a dynamic covalent polymer gel. In this approach, low molecular weight gelators self-assemble to form a gel first. For further modification active functional groups should be available. A crosslinking reagent with at least two complimentary functional groups is then allowed to react and crosslink the low molecular weight gelators located on the gel matrix to form a dynamic covalent polymer.
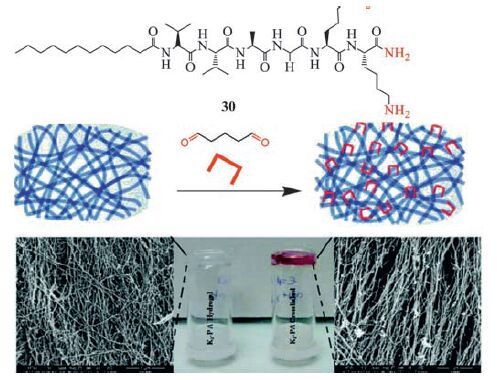
Post-modification of a peptide gel was reported by Guler and co-workers (Fig. 11) [114]. Peptide amphiphile molecules (Lauryl-VVAGKK-Am, 30) self-assemble into nanofibres with a diameter of ca. 10nm at pH> 7. A three-dimensional network of the nanofibres forms a self-supporting hydrogel. In the gelation, the lauryl group of 30 promotes hydrophobic interactions. Meanwhile, β-sheet forming amino acids facilitate hydrogen-bonding. The peptide gelator molecules possess amine functional groups, so glutaraldehyde can be used as crosslinking reagent to post-modify the peptide gel via imine bonding. The dynamic crosslinking process does not affect the nanofibre morphology, porosity and fibrous structure of the peptide gel. But it facilitates tuning and enhancing the viscoelastic characteristics (G’ ca. 80 KPa vs. 45 KPa) of the self-assembled peptide gels. This study shows that dynamic covalent bonding is able to stabilize low molecular weight physical gels formed by suparmolecular weak interactions.

|
Download:
|
| Figure 11. (a) Chemical structure of peptide amphiphile 30, (b) modification of peptide gel through a glutaraldehyde crosslinking strategy, (c) 30 gel (left) and crosslinked 30 peptide gel (right). Adapted with permission from [114]. Copyright © The Royal Society of Chemistry 2015. | |
{kind=link}
Polymerization of small molecules with multiple functional groups via dynamic covalent bonding yields dynamic covalent polymers. The molecular constituents are linked through dynamic covalent connections. For efficient gelation, dynamic covalent polymers are generally constructed from at least one multitopic monomer with three or more connection points. During the formation of dynamic covalent gels error correction through thermodynamic equilibration allows for efficient polymerization. Permanent microporosity may be achieved when the gel is constructed from comparatively rigid structural components [115]. Although dynamic covalent bonds are frequently used in the field of covalent-organic frameworks [116, 117], gels with rigid structural components have been developed only recently. Such gel materials are promising for storage, separation, delivery and catalysis. In such a class of gels formed by dynamic covalent polymers, dynamic covalent interactions are the main driving force to form the 3D gel network. Fascinatingly, although the small molecular precursors do not form gels, the resulting dynamic covalent polymers form. An infinite extended network structure is formed to gelate a large number of solvent molecules. Such gels of dynamic covalent polymers are generally boosted by gentle heating. They cannot be redissolved upon further heating and do not show thermoreversible gel-sol transitions.
When linear diamines react with dialdehydes, polycondensation occurs and 1D polyimine polymer is formed. Dynamic gel consisting of 1D imine polymer in the aid of metal-ligand coordination was reported by Nitschke and coworkers [118]. Subcomponent self-assembly of 1, 4-diaminobenzene and 4, 4'-diformyl-3, 3'-bipyridine induces gelation in DMSO in the presence of Cu+ templates (Fig. 12) . The gel matrix is constructed via the formation of dynamic covalent bonds around metal templates. Trioctylphosphine is introduced to cap the vacant coordination sites of Cu2+ ions. The imine metallopolymer undergoes unusual sol-to-gel transition on temperature rise that is reversed at room temperature. The photoluminescence of the gel system in DMSO solution is temperature dependent between 20 and 185 ℃. It allows for optical measurement of temperature determination or monitor. However, this is not a gel that is driven by dynamic covalent bonding, and instead crosslinking between 1D dynamic covalent polymer chains is achieved by metal-organic coordination.

|
Download:
|
| Figure 12. Schematic representation of gelation mechanism consisting of 1D imine polymer in the presence of Cu+ templates and photographs of inverted NMR tubes showing the sol-gel transition. Adapted with permission from [118]. Copyright © 2011 American Chemical Society. | |
{kind=link}
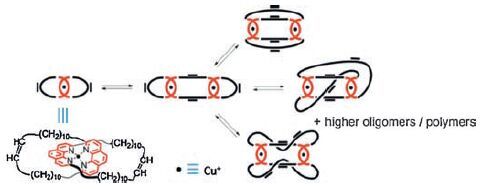
Interplay of dynamic covalent bonding and metal-organic coordination in gels has been demonstrated by Stefano and coworkers [119, 120]. A gel is obtained by ring opening metathesis polymerization of a copper(I) catenane (Fig. 13) [119]. The ringopening polymerization is achieved via olefin metathesis reaction in the aid of second generation Grubbs catalyst. The bis-(phenanthroline)copper(I) units are kept in the ring-opening. The ring-opening generates a mixture composed of oligomeric/ polymeric species of varying sizes. The resulting gel, mainly composed by interlocked species, is flexible and elastic. In addition, the gel can be easily reshaped and show reversible swelling. Besides dynamic olefin metathesis phenanthroline-Cu(I) coordination also plays an important role in the gelation because the gels do not form in the absence of metal. In a related example, after addition of Cu(I) in a dynamic library of phenanthroline containing cyclic molecules a gel is formed as well [120].

|
Download:
|
| Figure 13. Schematic representation of ring-opening olefin metathesis of Cu(I) catenane in the presence of second-generation grubbs catalyst. Adapted with permission from [119]. Copyright © 2015 American Chemical Society. | |
{kind=link}
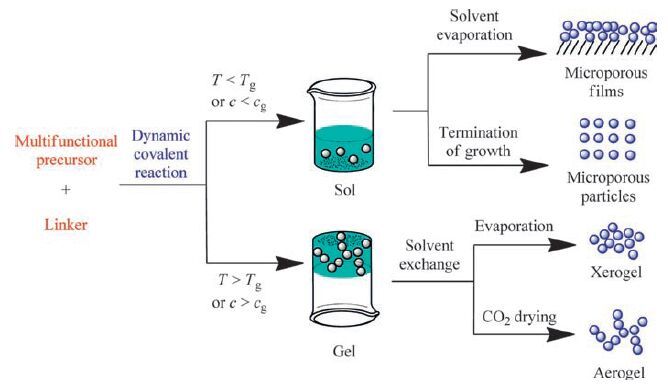
Gels that are driven by dynamic covalent bonding have been developed recently from low molecular weight molecules. Formation of 3D polymer network is generally believed to be responsible for the gelation and it requires that at least one monomer unit contain more than two dynamic functionalities. Like silicas, condensation of the precursors with multiple functional groups yields nanoparticles as a colloidal dispersion or a sol (Fig. 14) . Nanoparticles of microporous dynamic covalent polymer networks may be obtained if rigid building units are used. Solvent evaporation of the sol may result in formation of microporous films. Microporous nanoparticles can be obtained upon termination of particle growth. If these nanoparticles are interconnected to form a 3D matrix with hierarchically porous structure, a dynamic covalent gel may be obtained. The gel can be processed to yield a xerogel via solvent evaporation or an aerogel via subcritical or supercritical CO2 drying.

|
Download:
|
| Figure 14. Syntheses of nanoparticles, films, gels (xerogels and aerogels) from dynamic covalent reaction of small molecules. | |
{kind=link}
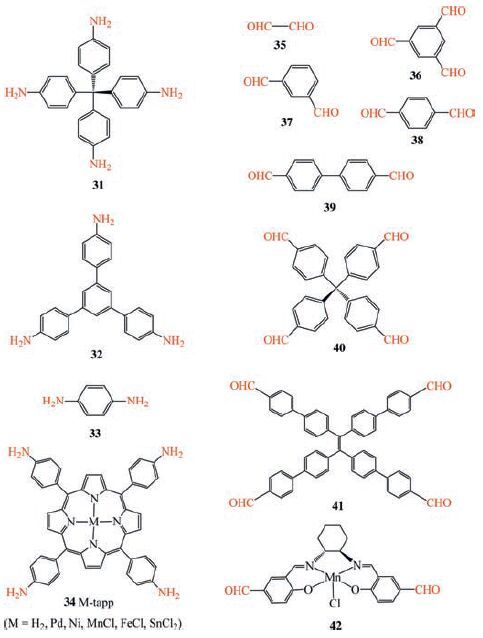
Polyimines from various bridging amines (31-34) and aldehydes (35-42) (Scheme 6) have been studied for gelation [121-123]. Gelation is achieved by condensation of rigid bridging amines and aldehydes under mild solvothermal conditions (80 ℃). The gels are obtained in water or in organic solvents (e.g., DMSO). The initially formed nanoparticles evidenced by dynamic light scattering (DLS) are held together by imine bonds to yield 3D crosslinked gel matrix. The unique sponge-like morphology based on microporous nanoparticles make it possible to get highly porous aerogels. Imine aerogels have been prepared from the imine gels by exchanging the solvent with subcritical CO2. The areogels show high porosity. The 31-35 imine aerogel prepared from tetrakis(4-aminophenyl)methane (31) and biformyl (35) exhibits the highest BET surface areas up to 1021 m2 g-1 (Fig. 15) .

|
Download:
|
| Scheme6. Various bridging amines (31-34) and aldehydes (35-42) that have been utilized for gelation study. | |
{kind=link}

|
Download:
|
| Figure 15. (a) SEM and (b) TEM micrographs of the 31-35 aerogel (the bars represent 300 and 50 nm for SEM and TEM, respectively). | |
{kind=link}
In comparison, imine aerogels have higher surface areas than the corresponding amorphous microporous networks [124], but lower than the corresponding crystalline covalent organic frameworks (COFs) with highly regular micropores [125]. As a general feature of aerogels, the imine aerogels show high pore volumes up to 2.97 cm3 g-1, which is much higher than COFs. Additionally the areogles exhibit hierarchical pore systems with a broad distribution of pore sizes. The pore sizes are tunable by simply varying the constituent molecular units. Therefore these imine aerogels are a novel class of porous materials showing higher thermal and hydrolytic stability, low densities and exclusion of expensive, toxic or reactive metals. In addition, the iminecrosslinked gel matrix can be disassembled by treatment with acid because benzoic imine bond is dynamic and is hydrolyzed under strongly acidic conditions. This suggests that the imine aerogels are readily degradable and regenerated.
High porosity of the imine aerogels makes them candidates as absorbents. They have been used in selective capture of CO2 because their pores are decorated with imine bonds and residue amino groups. The 31-35 aerogel possesses a CO2 uptake of 1.5 μmol g-1 at 298 K and 1.0 bar with the isosteric heat 38.1 kJ mol-1, and displays high CO2/N2 selectivity up to 70.9, which is derived from the ideal adsorbed solution theory [121].
The imine gels are readily functionalized by rational design of precursors with complementary reactive functional groups. Porphyrin functional units are incorporated into imine gels to get a series of functional porphyrin imine gels by reacting 5, 10, 15, 20-tetrakis(p-aminophenyl)porphyrin and its metalated derivatives (M-tapp, M = Pd, Ni, MnCl, FeCl, SnCl2) 34 with di/ multifunctional aldehydes (e.g., tetrakis-(4-formylphenyl)-methane, 40) [126]. Impregnation of metal ions in porphyrinbased imine gels is able to modulate guest uptake. The impregnation of various metal ions enhances the uptake capacity of various gases (CO2, H2, C2H4) despite higher densities of the aerogels. Among the metal ions, Pd(II) is the best to increase the CO2 adsorption capacity (1.62 μmol g-1, 7.13 wt% at 298 K, 1 bar) and the isosteric heat of CO2 adsorption (40.0 kJ mol-1) .
Aggregation-induced emission (AIE)-based luminescent imine gels have been developed with a tetraphenylethylene (TPE)-based tetraaldehyde precursor 41 [122]. AIE-based materials show potential applications in light-emitting diodes, fluorescence labeling, light harvesting, photonic devices, and so forth [127]. Among AIE compounds, TPE-based luminophores are of particular interest due to their typical AIE features, facile synthesis and ready fictionalization [128]. The TPE-based imine gel is synthesized via the reaction of 41 with a tetraamine 31. The resulting 31-41 gel/aerogel materials show interesting guest inclusion behaviors and aggregation-induced emission, and can be potentially utilized as chemosensors for explosives.
Incorporation of catalytically active centres into imine gels has been demonstrated by rational design of precursors. Such a dynamic covalent gel strategy has been successfully employed to immobilize an asymmetric Mn-salen catalyst within the channels of a microfluidic flow reactor (Fig. 16) [123]. The Mn-salen imine gel is synthesized via the reaction of a Mn-salen dialdehyde 42 with a tetraamine 31 and attached to the capillary surface. The surface of the inner wall of the capillaries is modified in advance with amine functional groups in order to anchor the Mn-salen imine gel via imine bonding. The Mn-salen imine gel is readily introduced into a fused-silica capillary reactor via in-situ gelation. The gel-capillary reactor have been proven effective for the enantioselective kinetic resolution of secondary alcohols (Scheme 8a), giving good conversions and enantiomeric excess (ee) (8 examples, conversions 46%-65%, ee 35%-91%). Under the gelcapillary flow reaction conditions optimal conversions are achieved in much shorter reaction times (15 min) compared to the batch process (120 min). In addition, the capillary reactor can be reused at least eight times without loss of activity or enantioselectivity.

|
Download:
|
| Figure 16. Schematic representation of the setup of a catalytic chiral gel microfluidic reactor using a capillary for asymmetric kinetic resolution of secondary alcohols, and SEM images of a cross-section of the capillary coated with Mn-salen gel prepared from 31 and 42 with about 2 μm thickness. | |
{kind=link}

|
Download:
|
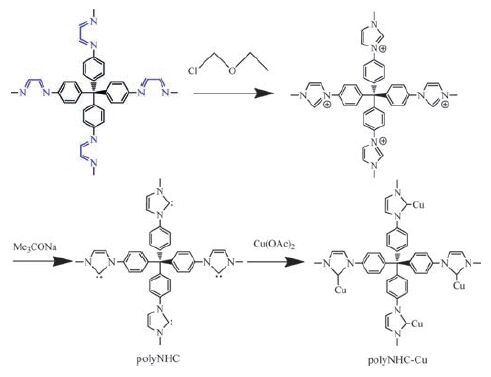
| Scheme7. Post-modification of the 31-35 imine gel to form polyNHC and polyNHCCu networks. | |
{kind=link}

|
Download:
|
| Scheme8. Catalytic reactions: (a) Enantioselective kinetic resolution of secondary alcohols catalyzed by Mn-salen gel prepared from 31 and 42, (b) Reaction of CO2 with epichlorohydrin to cyclic carbonate catalyzed by polyNHC, and (c) Deoxygenation of sulfoxides to sulfides with PhSiH3 catalyzed by polyNHC-Cu. | |
{kind=link}
The imine gels can be post-modified to incorporate catalytic centres. The 31-35 imine gel is converted into polyimidazolium chloride, then to a polyNHC (N-heterocyclic carbene) network by deprotonation, and finally to a Cu(II)-coordinated polyNHC network (Scheme 7) [129]. The catalytic property of polyNHC has been shown in the reaction of CO2 with epoxides to cyclic carbonates (epichlorohydrin, 94% yield) (Scheme 8b). The catalytic activity of polyNHC-Cu has been explored in the deoxygenation of sulfoxides with PhSiH3 as reductant (Scheme 8c). Various sulfoxides including aromatic and aliphatic sulfoxides were examined to yield the corresponding sulfides (7 examples, yields 11% to >99%). The stability of the catalyst was shown in further recycling steps and no significant loss in activity was observed for three cycles at least.
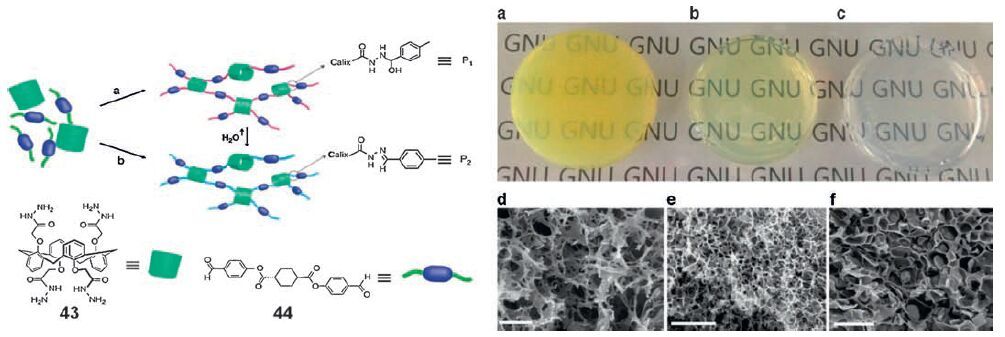
Ductile and high tensile strength calix[4]arene-derived gels have been produced via hydrazone linkage (Fig. 17) [130]. The gels are prepared from calix[4]arene-derivative 43 bearing four hydrazide groups and diphenyl terephthalate-devivative 44 bearing two aldehyde groups in DMSO. The hydrazide groups and the aldehyde groups react to form hydrazone bonding. Reversible sol-gel transition of the gel suggests that the resulting gel consists of discrete multi-meric gelator species rather than extended polymer structure. Interestingly the mechanical and ductile properties of the gels can be controlled by catalytic HCl. The tensile strength of the gel formed without HCl approaches over 40 MPa. The most remarkable aspect of this study is the mechanical property of the hydrogel. After the solvent DMSO was exchanged with water, the resulting hydrogel showed 7000-to 100, 000-fold enhanced mechanical properties. The hydrazone reaction produces-OH groups under non-acidic conditions or with addition of water, offering enhanced intermolecular hydrogenbonding interactions and thus increased elastic properties. This represents an interesting molding method from small molecules to retain the gel shape after processing. Good ionic conductivities as gel electrolytes were observed, which are on par with polymer gel type electrolytes.

|
Download:
|
| Figure 17. Schematic of gel formation by hydrazone reaction, (a) without addition of acid and (b) with addition of acid. Photograph of organogels produced using a mixture of 43 and 44 (42-84 μmol) in DMSO and SEM images of corresponding xerogels at different concentration of HCl. (a, d) 10 nmol HCl (after 24 h), (b, e) 5 nmol HCl (after 24 h), (c, f) without addition of HCl (after 36 h). Scale bars, 2 μm for d, e and 50 μm for f. | |
{kind=link}
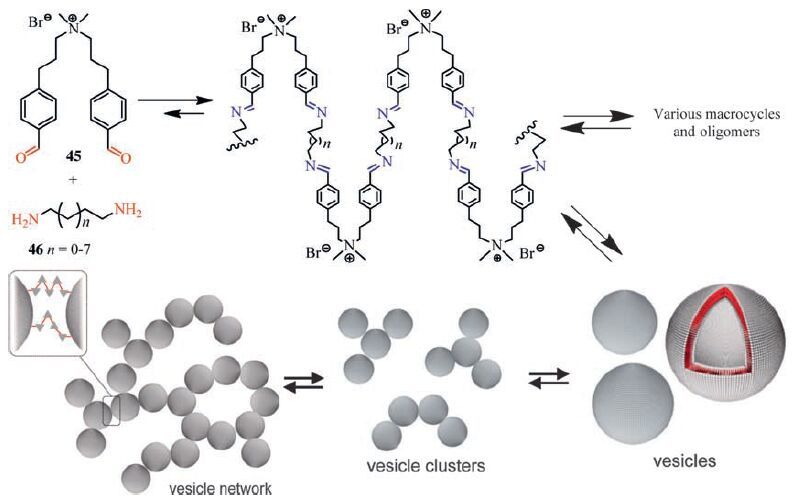
In the above examples, 3D extended polymer networks are formed to result in particle-based sponge-like morphology when relatively rigid or bulky building units are used. In contrast, when flexible building units are used, the building units may be folded and other morphologies may be obtained. Hydrogels from watersoluble cationic bisaldehyde 45 and 1, ω-alkyl bisamines (46) were reported by Van Esch and coworkers (Fig. 18) [131]. The hydrogels consist of 3D networks of connected vesicles. Initially flexible bisaldehyde react with bisamines to form vesicles [132], which reversibly crosslink to form a percolating vesicle network, leading to gelation of solvent molecules. Owing to the reversible nature of the imine bond, these vesicle gel assemblies are highly responsive to changes in pH and temperature.

|
Download:
|
| Figure 18. Formation of vesicle hydrogels from flexible bisaldehyde and bisamines via imine bonding. | |
{kind=link}
6. Conclusions and outlooks
Dynamic covalent chemistry has attracted particular research interest due to their potential applications in stimuli-responsive materials. It combines the robustness of covalent bonds with the flexibility typical for non-covalent interactions. In this review, we have summarized the advances in the gels with dynamic covalent bonding. The progress in suparmolecular and polymer gels with dynamic covalent bonding is described. Their structure-property relationship is analyzed. Rationally designed gelators have shown remarkable ability to generate novel functional materials.
The examples show that dynamic covalent gels are smart as suparmolecular gels in response to external stimuli. In the meantime, dynamic covalent gels are more stable than common supramolecular gels due to the strong nature of reversible covalent bonds. In situ rapid gelation has been controllably achieved (in the aid of catalyst) under ambient conditions based on dynamic covalent chemistry. The gelation does not need a heating-cooling process. This makes the gels applicable in some thermosensitive systems. Syntheses of dynamic covalent gels from small molecules offer a great many of possibilities to develop functional materials due to a wide range of structural variations and the easy synthetic accessibility of these small molecule precursors. It allows for rational design of building blocks with complementary functional groups. The porous structure and functionality of gels can thus be facilely tuned with the building units.
Despite the development in dynamic covalent reactions, dynamic covalent reactions applied for gels are still limited. There is still a lot of room for further developments. Development of more dynamic covalent reactions is also important to expand the synthetic scope of gels, e.g., urea formation [133-139]. Especially new reactions involving more robust bonds in the presence of catalyst are preferred to construct functional gels under thermodynamic control. New functional gel materials are expectable based on the dynamic covalent chemistry. Furthermore developing functional systems based on more than one type of dynamic covalent bonds remains a challenge to achieve multiple functionalities [140].
AcknowledgmentWe acknowledge the NSFC (Nos. 51573216 and 21273007) , the Program for New Century Excellent Talents in University (No.NCET-13-0615) and the FRF for the Central Universities (No.16lgjc66) for support.
| [1] | K. Almdal, J. Dyre, S. Hvidt, O. Kramer, Towards a phenomenological definition of the term 'gel'. Polym. Gels Netw. 1 (1993) 5–17. DOI:10.1016/0966-7822(93)90020-I |
| [2] | P.J. Flory, Introductory lecture. Faraday Discuss. Chem. Soc. 57 (1974) 7–18. DOI:10.1039/dc9745700007 |
| [3] | R.G. Weiss, P. Té rech, Molecular gels, in:R.G. Weiss, P. Té rech (Eds.), Materials With Self-assembled Fibrillar Networks, Springer, Netherlands, 2006, pp. 1-13. |
| [4] | J.M. Guenet, Microfibrillar networks:polymer thermoreversible gels vs organogels. Macromol. Symp. 241 (2006) 45–50. DOI:10.1002/(ISSN)1521-3900 |
| [5] | E. Dickinson, Structure and rheology of colloidal particle gels:insight from computer simulation, Adv. Colloid Interface Sci. 199-200(2013) 114-127. |
| [6] | Z.J. Zhao, J.W.Y. Lam, B.Z. Tang, Self-assembly of organic luminophores with gelation-enhanced emission characteristics. Soft Matter 9 (2013) 4564–4579. DOI:10.1039/c3sm27969c |
| [7] | J.M. Lehn, Supramolecular chemistry-scope and perspectives molecules, supermolecules, and molecular devices (Nobel Lecture). Angew. Chem. Int. Ed. 27 (1988) 89–112. DOI:10.1002/(ISSN)1521-3773 |
| [8] | J.M. Lehn, Perspectives in supramolecular chemistry-from molecular recognition towards molecular information processing and self-organization. Angew. Chem. Int. Ed. 29 (1990) 1304–1319. DOI:10.1002/(ISSN)1521-3773 |
| [9] | N.M. Sangeetha, U. Maitra, Supramolecular gels:functions and uses. Chem. Soc. Rev. 34 (2005) 821–836. DOI:10.1039/b417081b |
| [10] | P. Dastidar, Supramolecular gelling agents:can they be designed. Chem. Soc. Rev. 37 (2008) 2699–2715. DOI:10.1039/b807346e |
| [11] | E.A. Appel, F. Biedermann, U. Rauwald, Supramolecular cross-linked networks via host-guest complexation with cucurbit. J. Am. Chem. Soc. 132 (2010) 14251–14260. DOI:10.1021/ja106362w |
| [12] | S.Y. Dong, J.Y. Yuan, F.H. Huang, A pillar[5] arene/imidazolium[2] rotaxane:solvent-and thermo-driven molecular motions and supramolecular gel formation, Chem. Sci. 5(2014) 247-252. |
| [13] | Y.K. Tian, Y.G. Shi, Z.S. Yang, F. Wang, Responsive supramolecular polymers based on the bis[alkynylplatinum(II)] terpyridine molecular tweezer/arene recognition motif, Angew. Chem. Int. Ed. 53(2014) 6090-6094. |
| [14] | J.M. Hu, S.Y. Liu, Engineering responsive polymer building blocks with hostguest molecular recognition for functional applications. Acc. Chem. Res. 47 (2014) 2084–2095. DOI:10.1021/ar5001007 |
| [15] | S. Bhattacharjee, S. Bhattacharya, Charge transfer induces formation of stimuliresponsive, chiral, cohesive vesicles-on-α-string that eventually turn into a hydrogel. Chem. Asian J. 10 (2015) 572–580. DOI:10.1002/asia.201403205 |
| [16] | A. Das, S. Ghosh, Supramolecular assemblies by charge-transfer interactions between donor and acceptor chromophores. Angew. Chem. Int. Ed. 53 (2014) 2038–2054. DOI:10.1002/anie.201307756 |
| [17] | J.F. Xu, Y.Z. Chen, D.Y. Wu, Photoresponsive hydrogen-bonded supramolecular polymers based on a stiff stilbene unit. Angew. Chem. Int. Ed. 52 (2013) 9738–9742. DOI:10.1002/anie.201303496 |
| [18] | C. Rest, M.J. Mayoral, K. Fucke, Self-assembly and (hydro)gelation triggered by cooperative π-π and unconventional C-H…X hydrogen bonding interactions. Angew. Chem. Int. Ed. 53 (2014) 700–705. DOI:10.1002/anie.201307806 |
| [19] | K. Hanabusa, T. Miki, Y. Taguchi, T. Koyama, H. Shirai, Two-component, small molecule gelling agents, J. Chem. Soc. Chem. Commun. (1993) 1382-1384. |
| [20] | N.N. Adarsh, D.K. Kumar, P. Dastidar, Composites of N, N'-bis-(pyridyl) ureadicarboxylic acid as new hydrogelators-a crystal engineering approach. Tetrahedron 63 (2007) 7386–7396. DOI:10.1016/j.tet.2007.02.005 |
| [21] | L. Meazza, J.A. Foster, K. Fucke, Halogen-bonding-triggered supramolecular gel formation. Nat. Chem. 5 (2013) 42–47. |
| [22] | J.P. Hill, W.S. Jin, A. Kosaka, Self-assembled hexa-peri-hexabenzocoronene graphitic nanotube. Science 304 (2004) 1481–1483. DOI:10.1126/science.1097789 |
| [23] | Y. Feng, Z.T. Liu, J. Liu, Peripherally dimethyl isophthalate-functionalized poly(benzyl ether) dendrons:a new kind of unprecedented highly efficient organogelators. J. Am. Chem. Soc. 131 (2009) 7950–7951. DOI:10.1021/ja901501j |
| [24] | A.P. Sivadas, N.S.S. Kumar, D.D. Prabhu, Supergelation via purely aromatic π-π driven self-assembly of pseudodiscotic oxadiazole mesogens. J. Am. Chem. Soc. 136 (2014) 5416–5423. DOI:10.1021/ja500607d |
| [25] | T. Naota, H. Koori, Molecules that assemble by sound:an application to the instant gelation of stable organic fluids. J. Am. Chem. Soc. 127 (2005) 9324–9325. DOI:10.1021/ja050809h |
| [26] | D.J. Abdallah, S.A. Sirchio, R.G. Weiss, Hexatriacontane organogels. The first determination of the conformation and molecular packing of a low-molecular-mass organogelator in its gelled state. Langmuir 16 (2000) 7558–7561. DOI:10.1021/la000730k |
| [27] | L.Y. Gao, B. Zheng, Y. Yao, F.H. Huang, Responsive reverse giant vesicles and gel from self-organization of a bolaamphiphilic pillar[5] arene, Soft Matter 9(2013) 7314-7319. |
| [28] | X.Z. Yan, S.J. Li, T.R. Cook, Hierarchical self-assembly:well-defined supramolecular nanostructures and metallohydrogels via amphiphilic discrete organoplatinum(II) metallacycles. J. Am. Chem. Soc. 135 (2013) 14036–14039. DOI:10.1021/ja406877b |
| [29] | S.Y. Dong, Y. Luo, X.Z. Yan, et al., A dual-responsive supramolecular polymer gel formed by crown ether based molecular recognition, Angew. Chem. Int. Ed. 50(2011) 1905-1909. |
| [30] | S.Y. Dong, B. Zheng, D.H. Xu, et al., A crown ether appended super gelator with multiple stimulus responsiveness, Adv. Mater. 24(2012) 3191-3195. |
| [31] | Z.H. Qi, C.A. Schalley, Exploring macrocycles in functional supramolecular gels:from stimuli responsiveness to systems chemistry. Acc. Chem. Res. 47 (2014) 2222–2233. DOI:10.1021/ar500193z |
| [32] | X.Z. Yan, D.H. Xu, X.D. Chi, et al., A multiresponsive, shape-persistent, and elastic supramolecular polymer network gel constructed by orthogonal self-assembly, Adv. Mater. 24(2012) 362-369. |
| [33] | X.Z. Yan, T.R. Cook, J.B. Pollock, Responsive supramolecular polymer metallogel constructed by orthogonal coordination-driven self-assembly and host/guest interactions. J. Am. Chem. Soc. 136 (2014) 4460–4463. DOI:10.1021/ja412249k |
| [34] | Q.G. Wang, J.L. Mynar, M. Yoshida, High-water-contentmouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 463 (2010) 339–343. DOI:10.1038/nature08693 |
| [35] | L.E. Buerkle, S.J. Rowan, Supramolecular gels formed from multi-component low molecular weight species. Chem. Soc. Rev. 41 (2012) 6089–6102. DOI:10.1039/c2cs35106d |
| [36] | J. Raeburn, D.J. Adams, Multicomponent low molecular weight gelators. Chem. Commun. 51 (2015) 5170–5180. DOI:10.1039/C4CC08626K |
| [37] | A.R. Hirst, D.K. Smith, Two-component gel-phase materials-highly tunable selfassembling systems. Chem. Eur. J. 11 (2005) 5496–5508. DOI:10.1002/(ISSN)1521-3765 |
| [38] | J.Y. Zhang, C.Y. Su, Metal-organic gels:from discrete metallogelators to coordination polymers. Coord. Chem. Rev. 257 (2013) 1373–1408. DOI:10.1016/j.ccr.2013.01.005 |
| [39] | A.Y.Y. Tam, V.W.W. Yam, Recent advances in metallogels. Chem. Soc. Rev. 42 (2013) 1540–1567. DOI:10.1039/c2cs35354g |
| [40] | M.O.M. Piepenbrock, G.O. Lloyd, N. Clarke, J.W. Steed, Metal-and anion-binding supramolecular gels. Chem. Rev. 110 (2010) 1960–2004. DOI:10.1021/cr9003067 |
| [41] | R.J. Wojtecki, M.A. Meador, S.J. Rowan, Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 10 (2011) 14–27. DOI:10.1038/nmat2891 |
| [42] | J.M. Lehn, Dynamic combinatorial chemistry and virtual combinatorial libraries. Chem. Eur. J. 5 (1999) 2455–2463. DOI:10.1002/(ISSN)1521-3765 |
| [43] | P.T. Corbett, J. Leclaire, L. Vial, Dynamic combinatorial chemistry. Chem. Rev. 106 (2006) 3652–3711. DOI:10.1021/cr020452p |
| [44] | S.J. Rowan, S.J. Cantrill, G.R.L. Cousins, J.K.M. Sanders, J.F. Stoddart, Dynamic covalent chemistry. Angew. Chem. Int. Ed. 41 (2002) 898–952. DOI:10.1002/1521-3773(20020315)41:6<>1.0.CO;2-R |
| [45] | A. Herrmann, Dynamic combinatorial/covalent chemistry:a tool to read, generate and modulate the bioactivity of compounds and compound mixtures. Chem. Soc. Rev. 43 (2014) 1899–1933. DOI:10.1039/C3CS60336A |
| [46] | R.A.R. Hunta, S. Otto, Dynamic combinatorial libraries:new opportunities in systems chemistry. Chem. Commun. 47 (2011) 847–858. DOI:10.1039/C0CC03759A |
| [47] | E. Moulin, G. Cormos, N. Giuseppone, Dynamic combinatorial chemistry as a tool for the design of functional materials and devices. Chem. Soc. Rev. 41 (2012) 1031–1049. DOI:10.1039/C1CS15185A |
| [48] | J.W. Li, P. Nowak, S. Otto, Dynamic combinatorial libraries:from exploring molecular recognition to systems chemistry. J. Am. Chem. Soc. 135 (2013) 9222–9239. DOI:10.1021/ja402586c |
| [49] | D. Beaudoin, T. Maris, J.D. Wuest, Constructing monocrystalline covalent organic networks by polymerization. Nat. Chem. 5 (2013) 830–834. DOI:10.1038/nchem.1730 |
| [50] | P. Terech, R.G. Weiss, Low molecular mass gelators of organic liquids and the properties of their gels. Chem. Rev. 97 (1997) 3133–3160. DOI:10.1021/cr9700282 |
| [51] | L.A. Estroff, A.D. Hamilton, Water gelation by small organic molecules. Chem. Rev. 104 (2004) 1201–1218. DOI:10.1021/cr0302049 |
| [52] | A.R. Hirst, B. Escuder, J.F. Miravet, D.K. Smith, High-tech applications of selfassembling supramolecular nanostructured gel-phase materials:from regenerative medicine to electronic devices. Angew. Chem. Int. Ed. 47 (2008) 8002–8018. DOI:10.1002/anie.v47:42 |
| [53] | S.S. Babu, V.K. Praveen, A. Ajayaghosh, Functional π-gelators and their applications. Chem. Rev. 114 (2014) 1973–2129. DOI:10.1021/cr400195e |
| [54] | G.C. Yu, X.Z. Yan, C.Y. Han, F.H. Huang, Characterization of supramolecular gels. Chem. Soc. Rev. 42 (2013) 6697–6722. DOI:10.1039/c3cs60080g |
| [55] | T. Tu, W.W. Fang, Z.M. Sun, Visual-size molecular recognition based on gels. Adv. Mater. 25 (2013) 5304–5313. DOI:10.1002/adma.201301914 |
| [56] | M. Suzuki, K. Hanabusa, Polymer organogelators that make supramolecular organogels through physical cross-linking and self-assembly. Chem. Soc. Rev. 39 (2010) 455–463. DOI:10.1039/B910604A |
| [57] | C.J. Kloxin, C.N. Bowman, Covalent adaptable networks:smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 42 (2013) 7161–7173. DOI:10.1039/C3CS60046G |
| [58] | H.Y. Wang, S.C. Heilshorn, Adaptable hydrogel networks with reversible linkages for tissue engineering. Adv. Mater. 27 (2015) 3717–3736. DOI:10.1002/adma.v27.25 |
| [59] | Y.H. Jin, C. Yu, R.J. Denman, W. Zhang, Recent advances in dynamic covalent chemistry. Chem. Soc. Rev. 42 (2013) 6634–6654. DOI:10.1039/c3cs60044k |
| [60] | C.D. Meyer, C.S. Joiner, J.F. Stoddart, Template-directed synthesis employing reversible imine bond formation. Chem. Soc. Rev. 36 (2007) 1705–1723. DOI:10.1039/b513441m |
| [61] | M.L.C. Quan, D.J. Cram, Constrictive binding of large guests by a hemicarcerand containing four portals. J. Am. Chem. Soc. 113 (1991) 2754–2755. DOI:10.1021/ja00007a060 |
| [62] | T.T. Tidwell, Hugo (Ugo) schiff, schiff Bases, and a century of β-lactam synthesis. Angew. Chem. Int. Ed. 47 (2008) 1016–1020. DOI:10.1002/(ISSN)1521-3773 |
| [63] | M.E. Belowich, J.F. Stoddart, Dynamic imine chemistry. Chem. Soc. Rev. 41 (2012) 2003–2024. DOI:10.1039/c2cs15305j |
| [64] | M. Ciaccia, S.D. Stefano, Mechanisms of imine exchange reactions in organic solvents. Org. Biomol. Chem. 13 (2015) 646–654. DOI:10.1039/C4OB02110J |
| [65] | S. Yamada, Advancement in stereochemical aspects of schiff base metal complexes, Coord. Chem. Rev. 190-192(1999) 537-555. |
| [66] | M. Rezaeivala, H. Keypour, Schiff base and non-schiff base macrocyclic ligands and complexes incorporating the pyridine moiety-the first 50 years. Coord. Chem. Rev. 280 (2014) 203–253. DOI:10.1016/j.ccr.2014.06.007 |
| [67] | H. Vardhan, A. Mehta, I. Nath, F. Verpoort, Dynamic imine chemistry in metal-organic polyhedra. RSC Adv. 5 (2015) 67011–67030. DOI:10.1039/C5RA10801B |
| [68] | A. Dirksen, S. Dirksen, T.M. Hackeng, P.E. Dawson, Nucleophilic catalysis of hydrazone formation and transimination:implications for dynamic covalent chemistry. J. Am. Chem. Soc. 128 (2006) 15602–15603. DOI:10.1021/ja067189k |
| [69] | A. Sanyal, Diels-Alder cycloaddition-cycloreversion:a powerful combo in materials design. Macromol. Chem. Phys. 211 (2010) 1417–1425. DOI:10.1002/macp.v211:13 |
| [70] | X.X. Chen, M.A. Dam, K. Ono, et al., A thermally re-mendable cross-linked polymeric material, Science 295(2002) 1698-1702. |
| [71] | S.D. Bergman, F. Wudl, Mendable polymers. J. Mater. Chem. 18 (2008) 41–62. DOI:10.1039/B713953P |
| [72] | B.J. Adzima, H.A. Aguirre, C.J. Kloxin, T.F. Scott, C.N. Bowman, Rheological, chemical analysis of reverse gelation in a covalently cross-linked diels-alder polymer network. Macromolecules 41 (2008) 9112–9117. DOI:10.1021/ma801863d |
| [73] | A.J. Inglis, L. Nebhani, O. Altintas, F.G. Schmidt, C. Barner-Kowollik, Rapid bonding/debonding on demand:reversibly cross-linked functional polymers via diels-alder chemistry. Macromolecules 43 (2010) 5515–5520. DOI:10.1021/ma100945b |
| [74] | J.Q. Zhang, Y. Niu, C.L. Huang, Self-healable and recyclable triple-shape PPDO-PTMEG co-network constructed through thermoreversible Diels-Alder reaction. Polym. Chem. 3 (2012) 1390–1393. DOI:10.1039/c2py20028g |
| [75] | G.C. Vougioukalakis, R.H. Grubbs, Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 110 (2010) 1746–1787. DOI:10.1021/cr9002424 |
| [76] | R.R. Schrock, Recent advances in olefin metathesis by molybdenum and tungsten imido alkylidene complexes. J. Mol. Catal. A:Chem. 213 (2004) 21–30. DOI:10.1016/j.molcata.2003.10.060 |
| [77] | Y.X. Lu, F. Tournilhac, L. Leibler, Z.B. Guan, Making insoluble polymer networks malleable via olefin metathesis. J. Am. Chem. Soc. 134 (2012) 8424–8427. DOI:10.1021/ja303356z |
| [78] | K.D. Okochi, Y.H. Jin, W. Zhang, Highly efficient one-pot synthesis of heterosequenced shape-persistent macrocycles through orthogonal dynamic covalent chemistry (ODCC). Chem. Commun. 49 (2013) 4418–4420. DOI:10.1039/C2CC33078D |
| [79] | M.J. Marsella, H.D. Maynard, R.H. Grubbs, Template-directed ring-closing metathesis:synthesis and polymerization of unsaturated crown ether analogs. Angew. Chem. Int. Ed. 36 (1997) 1101–1103. DOI:10.1002/(ISSN)1521-3773 |
| [80] | H. Otsuka, T. Muta, M. Sakada, T. Maeda, A. Takahara, Scrambling reaction between polymers prepared by step-growth and chain-growth polymerizations:macromolecular cross-metathesis between 1, 4-polybutadiene and olefin-containing polyester, Chem. Commun. (2009) 1073-1075. |
| [81] | Y.H. Jin, A.B. Zhang, Y.S. Huang, W. Zhang, Shape-persistent arylenevinylene macrocycles (AVMs) prepared via acyclic diene metathesis macrocyclization (ADMAC). Chem. Commun. 46 (2010) 8258–8260. DOI:10.1039/c0cc02941f |
| [82] | R. Nishiyabu, Y. Kubo, T.D. James, J.S. Fossey, Boronic acid building blocks:tools for self assembly. Chem. Commun. 47 (2011) 1124–1150. DOI:10.1039/C0CC02921A |
| [83] | D.G. Hall, Rhodium-catalyzed additions of boronic acids to alkenes and carbonyl compounds, in:D.G. Hall (Ed.), Boronic Acids:Preparation and Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, 2005, pp. 1-100. |
| [84] | D. Witt, Recent developments in disulfide bond formation. Synthesis 16 (2008) 2491–2509. |
| [85] | H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda, A. Takahara, A dynamic covalent polymer driven by disulfide metathesis under photoirradiation, Chem. Commun. 46(2010) 1150-1152. |
| [86] | T. Maeda, H. Otsuka, A. Takahara, Dynamic covalent polymers:reorganizable polymers with dynamic covalent bonds. Prog. Polym. Sci. 34 (2009) 581–604. DOI:10.1016/j.progpolymsci.2009.03.001 |
| [87] | T.F. Scott, A.D. Schneider, W.D. Cook, C.N. Bowman, Photoinduced plasticity in cross-linked. Science 308 (2005) 1615–1617. DOI:10.1126/science.1110505 |
| [88] | D. Montarnal, M. Capelot, F. Tournilhac, L. Leibler, Silica-like malleable materials from permanent organic networks. Science 334 (2011) 965–968. DOI:10.1126/science.1212648 |
| [89] | S.D. Stefano, R. Cacciapaglia, L. Mandolini, Supramolecular control of reactivity and catalysis-effective molarities of recognition-mediated bimolecular reactions. Eur. J. Org. Chem. 2014 (2014) 7304–7315. DOI:10.1002/ejoc.v2014.33 |
| [90] | K.N. Long, The mechanics of network polymers with thermally reversible linkages. J. Mech. Phys. Solids 63 (2014) 386–411. DOI:10.1016/j.jmps.2013.08.017 |
| [91] | Z. Wei, J.H. Yang, J.X. Zhou, Self-healing gels based on constitutional dynamic chemistry and their potential applications. Chem. Soc. Rev. 43 (2014) 8114–8131. DOI:10.1039/C4CS00219A |
| [92] | N. Sreenivasachary, J.M. Lehn, Gelation-driven component selection in the generation of constitutional dynamic hydrogels based on guanine-quartet formation. Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 5938–5943. DOI:10.1073/pnas.0501663102 |
| [93] | J.S. Foster, J.M. Z·urek, N.M.S. Almeida, Gelation landscape engineering using a multi-reaction supramolecular hydrogelator system. J. Am. Chem. Soc. 137 (2015) 14236–14239. DOI:10.1021/jacs.5b06988 |
| [94] | J. Boekhoven, J.M. Poolman, C. Maity, Catalytic control over supramolecular gel formation. Nat. Chem. 5 (2013) 433–437. DOI:10.1038/nchem.1617 |
| [95] | R. Eelkema, J.H. van Esch, Catalytic control over the formation of supramolecular materials. Org. Biomol. Chem. 12 (2014) 6292–6296. DOI:10.1039/C4OB01108B |
| [96] | J.M. Poolman, C. Maity, J. Boekhoven, et al., A toolbox for controlling the properties and functionalisation of hydrazone-based supramolecular hydrogels, J. Mater. Chem. B 4(2016) 852-858. |
| [97] | H. Qian, I. Aprahamian, An emissive and pH switchable hydrazone-based hydrogel. Chem. Commun. 51 (2015) 11158–11161. DOI:10.1039/C5CC03007B |
| [98] | C.B. Minkenberg, L. Florusse, R. Eelkema, G.J.M. Kope, J.H. van Esch, Triggered self-assembly of simple dynamic covalent surfactants. J. Am. Chem. Soc. 131 (2009) 11274–11275. DOI:10.1021/ja902808q |
| [99] | G.T. Wang, J.B. Lin, X.K. Jiang, Z.T. Li, Cholesterol-appended aromatic imine organogelators:a case study of gelation-driven component selection. Langmuir 25 (2009) 8414–8418. DOI:10.1021/la804188z |
| [100] | K. Lv, L. Qin, X.F. Wang, L. Zhang, M.H. Liu, A chiroptical switch based on supramolecular chirality transfer through alkyl chain entanglement and dynamic covalent bonding, Phys. Chem. Chem. Phys. 15(2013) 20197-20202. |
| [101] | Q.X. Jin, L. Zhang, X.F. Zhu, P.F. Duan, M.H. Liu, Amphiphilic schiff base organogels:metal-ion-mediated chiral twists and chiral recognition. Chem. Eur. J. 18 (2012) 4916–4922. DOI:10.1002/chem.v18.16 |
| [102] | H. Bunzen, E. Nonappa, S. Kalenius, E. Hietala, Kolehmainen, subcomponent selfassembly:a quick way to new metallogels. Chem. Eur. J. 19 (2013) 12978–12981. DOI:10.1002/chem.v19.39 |
| [103] | L.B. Zang, H.X. Shang, D.Y. Wei, S.M. Jiang, A multi-stimuli-responsive organogel based on salicylidene Schiff base, Sens. Actuators B 185(2013) 389-397. |
| [104] | J.W. Li, J.M.A. Carnall, M.C.A. Stuart, S. Otto, Hydrogel formation upon photoinduced covalent capture of macrocycle stacks from dynamic combinatorial libraries. Angew. Chem. Int. Ed. 50 (2011) 8384–8386. DOI:10.1002/anie.v50.36 |
| [105] | L.J. Prins, P. Scrimin, Covalent capture:merging covalent and noncovalent synthesis. Angew. Chem. Int. Ed. 48 (2009) 2288–2306. DOI:10.1002/anie.200803583 |
| [106] | K. Sada, M. Takeuchi, N. Fujita, M. Numata, S. Shinkai, Post-polymerization of preorganized assemblies for creating shape-controlled functional materials. Chem. Soc. Rev. 36 (2007) 415–435. DOI:10.1039/B603555H |
| [107] | I.A. Coates, D.K. Smith, Controlled self-assembly-synthetic tunability and covalent capture of nanoscale gel morphologies. Chem. Eur. J. 15 (2009) 6340–6344. DOI:10.1002/chem.v15:26 |
| [108] | C.H. Ren, Z.J. Song, W.T. Zheng, Disulfide bond as a cleavable linker for molecular self-assembly and hydrogelation. Chem. Commun. 47 (2011) 1619–1621. DOI:10.1039/C0CC04135A |
| [109] | Y. Shi, J.Y. Wang, H.M. Wang, et al., Glutathione-triggered formation of a fmocprotected short peptide-based supramolecular hydrogel, PLOS ONE 9(2014) e106968. |
| [110] | R.J. Williams, A.M. Smith, R. Collins, Enzyme-assisted self-assembly under thermodynamic control. Nat. Nanotechnol. 4 (2009) 19–24. DOI:10.1038/nnano.2008.378 |
| [111] | S. Toledano, R.J. Williams, V. Jayawarna, R.V. Ulijn, Enzyme-triggered selfassembly of peptide hydrogels via reversed hydrolysis. J. Am. Chem. Soc. 128 (2006) 1070–1071. DOI:10.1021/ja056549l |
| [112] | P. Kaur, J.T. Hupp, S.T. Nguyen, Porous organic polymers in catalysis:opportunities and challenges. ACS Catal. 1 (2011) 819–835. DOI:10.1021/cs200131g |
| [113] | Y.G. Zhang, S.N. Riduan, Functional porous organic polymers for heterogeneous catalysis. Chem. Soc. Rev. 41 (2012) 2083–2094. DOI:10.1039/C1CS15227K |
| [114] | M.A. Khalily, M. Goktas, M.O. Guler, Tuning viscoelastic properties of supramolecular peptide gels via dynamic covalent crosslinking. Org. Biomol. Chem. 13 (2015) 1983–1987. DOI:10.1039/C4OB02217C |
| [115] | N.B. McKeown, P.M. Budd, Exploitation of intrinsic microporosity in polymerbased materials. Macromolecules 43 (2010) 5163–5176. DOI:10.1021/ma1006396 |
| [116] | A.P. Côté, A.I. Benin, N.W. Ockwig, Porous, crystalline, covalent organic frameworks. Science 310 (2005) 1166–1170. DOI:10.1126/science.1120411 |
| [117] | S.Y. Ding, W. Wang, Covalent organic frameworks (COFs):from design to applications. Chem. Soc. Rev. 42 (2013) 548–568. DOI:10.1039/C2CS35072F |
| [118] | X. de Hatten, N. Bell, N. Yufa, G. Christmann, J.R. Nitschke, A dynamic covalent, luminescent metallopolymer that undergoes sol-to-gel transition on temperature rise, J. Am. Chem. Soc. 133(2011) 3158-3164. |
| [119] | J.A. Berrocal, L.M. Pitet, M.M.L. Nieuwenhuizen, et al., Ring-opening metathesis polymerization of a diolefinic[2]-catenane-copper(I) complex:an easy route to polycatenanes, Macromolecules 48(2015) 1358-1363. |
| [120] | J.A. Berrocal, S. Albano, L. Mandolini, S.D. Stefano, A CuI-based metallo-supramolecular gel-like material built from a library of oligomeric ligands featuring exotopic 1, 10-phenanthroline units, Eur. J. Org. Chem. 2015(2015) 7504-7510. |
| [121] | J.Y. Zhang, L.P. Liu, H.L. Liu, Highly porous aerogels based on imine chemistry:syntheses and sorption properties. J. Mater. Chem. A 3 (2015) 10990–10998. DOI:10.1039/C5TA00557D |
| [122] | W.J. Luo, Y.X. Zhu, J.Y. Zhang, et al., A dynamic covalent imine gel as a luminescent sensor, Chem. Commun. 50(2014) 11942-11945. |
| [123] | H.L. Liu, J. Feng, J.Y. Zhang, et al., A catalytic chiral gel microfluidic reactor assembled via dynamic covalent chemistry, Chem. Sci. 6(2015) 2292-2296. |
| [124] | P.A. Kerneghan, S.D. Halperin, D.L. Bryce, K.E. Maly, Postsynthetic modification of an imine-based microporous organic network. Can. J. Chem. 89 (2011) 577–582. DOI:10.1139/v11-014 |
| [125] | F.J. Uribe-Romo, J.R. Hunt, H. Furukawa, et al., A crystalline imine-linked 3-D porous covalent organic framework, J. Am. Chem. Soc. 131(2009) 4570-4571. |
| [126] | L.H. Zeng, P.S. Liao, H.L. Liu, Impregnation of metal ions into porphyrinbased imine gels to modulate guest uptake and to assemble a catalytic microfluidic reactor. J. Mater. Chem. A 4 (2016) 8328–8336. DOI:10.1039/C6TA01035K |
| [127] | J.D. Luo, Z.L. Xie, J.W.Y. Lam, et al., Aggregation-induced emission of 1-methyl-1, 2, 3, 4, 5-pentaphenylsilole, Chem. Commun. (2001) 1740-1741. |
| [128] | Z.J. Zhao, J.W.Y. Lam, B.Z. Tang, Tetraphenylethene:a versatile AIE building block for the construction of efficient luminescent materials for organic light-emitting diodes. J. Mater. Chem. 22 (2012) 23726–23740. DOI:10.1039/c2jm31949g |
| [129] | K. Thiel, R. Zehbe, J. Roeser, et al., A polymer analogous reaction for the formation of imidazolium and NHC based porous polymernetworks, Polym. Chem. 4(2013) 1848-1856. |
| [130] | J.H. Lee, J. Park, J.W. Park, et al., Supramolecular gels with high strength by tuning of calix[4] arene-derived networks, Nat. Commun. 6(2015) 6650. |
| [131] | C.B. Minkenberg, W.E. Hendriksen, F. Li, Dynamic covalent assembly of stimuli responsive vesicle gels. Chem. Commun. 48 (2012) 9837–9839. DOI:10.1039/c2cc34863b |
| [132] | C.B. Minkenberg, F. Li, P. van Rijn, Responsive vesicles from dynamic covalent surfactants. Angew. Chem. Int. Ed. 50 (2011) 3421–3424. DOI:10.1002/anie.201007401 |
| [133] | H.Z. Ying, Y.F. Zhang, J.J. Cheng, Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun. 5 (2014) 3218. |
| [134] | M. Hutchby, C.E. Houlden, J.G. Ford, Hindered ureas as masked isocyanates:facile carbamoylation of nucleophiles under neutral conditions. Angew. Chem. Int. Ed. 48 (2009) 8721–8724. DOI:10.1002/anie.v48:46 |
| [135] | E. Delebecq, J.P. Pascault, B. Boutevin, F. Ganachaud, On the versatility of urethane/urea bonds:reversibility, blocked isocyanate, and non-isocyanate polyurethane. Chem. Rev. 113 (2013) 80–118. DOI:10.1021/cr300195n |
| [136] | S.Y. Moon, J.S. Bae, E. Jeon, J.W. Park, Organic sol-gel synthesis:solution-processable microporous organic networks. Angew. Chem. Int. Ed. 49 (2010) 9504–9508. DOI:10.1002/anie.201002609 |
| [137] | S.Y. Moon, E. Jeon, J.S. Bae, M. Byeona, J.W. Park, Polyurea networks via organic sol-gel crosslinking polymerization of tetrafunctional amines and diisocyanates and their selective adsorption and filtration of carbon dioxide. Polym. Chem. 5 (2014) 1124–1131. DOI:10.1039/c3py01593a |
| [138] | S.Y. Moon, H.R. Mo, M.K. Ahn, Organic sol-gel synthesis of microporous molecular networks containing spirobifluorene and tetraphenylmethane nodes. J. Polym. Sci. A:Polym. Chem. 51 (2013) 1758–1766. DOI:10.1002/pola.26552 |
| [139] | S. Bhunia, N. Chatterjee, S. Das, K.D. Saha, A. Bhaumik, Porous polyurea network showing aggregation induced white light emission, applications as biosensor and scaffold for drug delivery. ACS Appl. Mater. Interfaces 6 (2014) 22569–22576. DOI:10.1021/am5066859 |
| [140] | A. Wilson, G. Gasparini, S. Matile, Functional systems with orthogonal dynamic covalent bonds. Chem. Soc. Rev. 43 (2014) 1948–1962. DOI:10.1039/C3CS60342C |