 2017, Vol. 28
2017, Vol. 28 



b Pony Testing International Group, Wuhan 430034, China;
c WuXi AppTec, Wuhan 430075, China;
d Hengshui University, Hengshui 053000, China;
e Synthetic and Functional Biomolecules Center, Peking University, Beijing 100871, China
Tuberculosis (TB) is one of the most common, communicable, and fatal diseases known to mankind. Millions of new cases occur every year throughout the world, and one-third of the world population is potentially infected with TB. Tuberculosis is caused by mycobacteria, predominantly Mycobacterium tuberculosis (MTB) [1]. The first-line drugs isoniazid (INH), rifampicin (RIF), pyrazinamide, and ethambutol are crucial therapeutics for the treatment of TB. Unfortunately, these drugs are becoming less effective due to the increasing prevalence of multi-drug-resistant TB (MDR-TB) and extensively drug-resistant TB. Therefore, there is an urgent need to develop novel, effective, and fast acting anti-TB drugs with low toxicity and activity against both actively growing and latent infections [1]. In the last 50 years, only a few drugs have been approved by the United States Food and Drug Administration for TB therapy, reflecting the inherent difficulties of developing new antiTB agents. Substantial effort needs to overcome these difficulties and meet the urgent need [2-40]. Currently, there are three main approaches to developing novel anti-TB agents [1, 2]: (1) Expanding the antibacterial spectrum by applying existing drugs to the treatment of TB. For example, ciprofloxacin (CPFX) is currently recommended as second-line agent by the World Health Organization for the treatment of TB, primarily involving resistance or intolerance to first-line anti-TB therapy [3]. (2) Searching for novel structures and mechanisms that have never been used to act against the TB organism, such as TMC 207 for the treatment of MDR-TB [38, 39]. (3) Synthesizing new analogs or modifying existing drug compounds that can shorten and improve TB treatment. One modern concept in drug design is molecular hybridization, which is based on combining the pharmacophore moieties of different bioactive substances to produce a new hybrid compounds with improved affinity and efficacy compared to the parent drugs [4]. Isatin is an important endogenous compound identified in many organisms [5] which has obtained considerable attention in medicinal chemistry due to its various biological activities such as anti-TB [6], anti-bacterial [7], anti-fungal [8], anti-virus, anti-tumor, anti-HIV [9], anti-inflammatory, analgesic, anti-convulsant, anti-viral, anthelmintic anti-oxidant, and central nervous system depressant (Fig. 1) . In addition, compounds with the isatin structural scaffold have been reported to be potent DNA gyrase inhibitors.

|
Download:
|
| Figure 1. Chemical structure of isatin. | |
{kind=link}
Isatin is a biologically versatile structure, and the stability of its indole nucleus has inspired medicinal chemists to introduce many pharmacophore moieties to obtain new potential anti-TB agents. Moreover, the structure-activity relationships (SAR) study of isatin derivatives revealed that 5-halogenation, N-alkylation, N-Mannich base and 3-thiosemicarbazone formation were effective in increasing inhibition activity against various bacteria, fungi, and viruses [10]. Therefore, isatin is a reasonable choice used to develop new anti-TB agents.
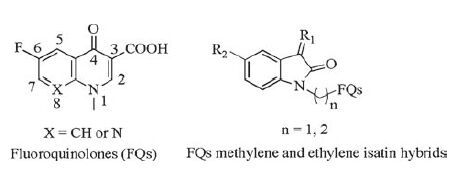
2. Isatin hybrids and their anti-tuberculosis activity 2.1. Isatin-quinolone/fluoroquinolone hybridsFluoroquinolones (FQs, Fig. 2) exhibited excellent antibacterial activity, and some of them are currently recommended as the second-line agents by the World Health Organization for the treatment of TB [3]. Moreover, the lipophilicity of the FQs plays an important role in the penetration of these compounds into bacterial cells, which suggests that increasing the lipophilic character at C-7 position could also increase the anti-TB activity [11]. Therefore, several series of FQs-isatin hybrids with remarkable improvement in lipophilicity have been synthesized and their anti-TB activity has been explored.

|
Download:
|
| Figure 2. Chemical structures of FQs and FQs methylene and ethylene isatin derivatives. | |
{kind=link}
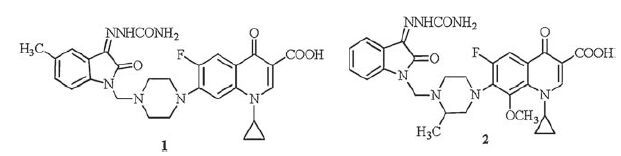
Three series of FQ methylene isatin hybrids (including norfloxacin, ciprofloxacin, and gatifloxacin) were synthesized and their anti-TB activity was evaluated by Sriram et al. [11-13] using the molecular hybridization drug design principle. The result indicated that these novel hybrids have potent activity against MTB. Among these FQs, the ciprofloxacin (CPFX) methylene isatin hybrid 1 (MIC: 1.39 nmol/L) was more active than moxifloxacin (MXFX, MIC: 1.94 nmol/L) and CPFX (MIC: 6.04 nmol/L) against MTB H37Rv (Fig. 3) . Its cell toxicity (IC50 > 111.29 nmol/L) was far less than the references (IC50 > 15.58 and >30.21 nmol/L for MXFX and CPFX, respectively). Hybrid 1 decreased the bacterial load in spleen tissue (mean colony forming units of 6.08) with 0.76-log 10 protections and was considered to be moderately active in reducing the bacterial count in the spleen. Unfortunately, in the lung tissue, hybrid 1 was found to be inactive. Gatifloxacin (GTFX) is in a phase III clinical trial for treatment of TB, so GTFX is also a candidate for structure modification. GTFX methylene isatin hybrid 2 was found to be 16 and 64 fold more potent than the parent GTFX against MTB and MDR-TB, while hybrid 2 (IC50: 3.0 μg/mL) was also found to be slightly more active than GTFX in the inhibition of the supercoiling activity of wild-type MTB DNA gyrase with dosedependent inhibition (Fig. 3) . The toxicity of hybrid 2 (IC50: 62.5 μg/mL) in a mammalian VERO cell line was very low, and the selectivity index (IC50/MIC) was more than 1250; in an in vivo mouse model (infected with MTB ATCC35801, 50 mg/kg), hybrid 2 decreased the bacterial load in the lung and spleen tissues with 3.62-and 3.76-log 10 protections, respectively [13].

|
Download:
|
| Figure 3. Ciprofloxacin methylene isatin 1 and gatifloxacin methylene isatin 2. | |
{kind=link}
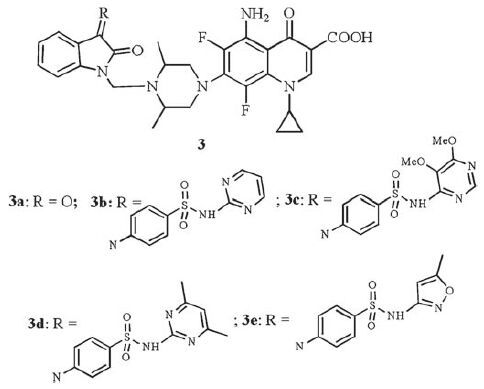
Mannich bases of sparfloxacin-isatin hybrids 3 were evaluated for the anti-TB activity [14] (Fig. 4) . All of the targets showed potent activity against MTB H37Rv (MIC: <6.25 μg/mL). However, they are all less active than the parent sparfloxacin (MIC: 0.5 μg/mL).
SAR indicated that C-8 methyloxy FQ derivatives with N1-cyclopropyl substitution are not only much more active against resistant MTB than corresponding C-8 hydrogen compounds, but are also able to decrease the possibility of drug-resistant MTB [15]. On the basis of the SAR results, a series of 1-cyclopropyl-8-methyloxy FQs (GTFX, balofloxacin/BLFX, 8-OCH3 CPFX, and MXFX) methylene/ethylene isatin hybrids were designed, synthesized, and evaluated their in vitro antimycobacterial activity by our group [1, 16-18] (Fig. 5) . The preliminary results indicated that all of the FQ ethylene isatin hybrids exhibited moderate to excellent anti-TB activity: BLFX ethylene isatin hybrid 4 revealed most active potency against MTB H37Rv ATCC 27294 and MDR-MTB 09710 (MIC: 0.25-< 0.5 μg/mL), which was comparable to MXFA and 32-fold more potent than BLFX. Regarding 8-OCH3 CPFX mannich bases, all of the synthesized methylene/ethylene isatin hybrids were less active than the parent 8-OCH3 CPFX against Mycobacterium smegmatis CMCC 93202, but most of the methylene isatin derivatives were more active than 8-OCH3 CPFX, CPFX, INH, and RIF against MTB H37Rv ATCC 27294. It was noted that compound 5a (MIC: 0.074 μmol/L) was 2-13 fold more potent than the reference compounds 8-OCH3 CPFX, CPFX, INH, and RIF (MIC: 0.19-0.97 μmol/L) against MTB H37Rv ATCC 27294, and compound 5b (MIC: 6.72 μmol/L) was about 1.6 times more potent than the parent 8-OCH3 CPFX and 3.5 times more potent than CPFX against MDR-MTB 09710. MXFX methylene/ethylene isatin hybrids were also found to have considerable antimycobacterial activity (MIC: 4-> 16 μg/mL). Among them, the most active MXFX methylene isatin hybrids, 6a and 6b, were 2-64 fold more active than INH and RIF against M. smegmatis CMCC 93202, 2 fold more than RIF against MTB H37Rv ATCC 27294, and 16-> 64 times greater than INH, RIF, and ethambutol against MDR-MTB 09710 isolate. The above results demonstrated that the lipophilicity of the tested compounds was not the sole parameter affecting antimycobacterial activity.

|
Download:
|
| Figure 4. Mannich bases of sparfloxacin-isatin hybrids 3. | |
{kind=link}

|
Download:
|
| Figure 5. 1-Cyclopropyl-8-methyloxy FQs methylene/ethylene isatin hybrids 4-6. | |
{kind=link}
A series of heteryl subtstituted isatins nalidixic acid Schiff bases 7 were screened for their in vitro antimycobacterial activity against Mycobacterium intercellulari, Mycobacterium xenopi, Mycobacterium cheleneo, and M. smegmatis [2] (Fig. 6) . 7f displayed potent anti-TB activity (MIC: 0.625 μg/mL), which is 20-fold more potent than the reference drug INH (MIC: 12.5 μg/mL), while only modest anti-TB activity was observed for the rest compounds. The pharmacophoric model that was built based on these results revealed the necessity of the following pharmacophoric features for anti-TB activity: aromatic center, hydrogen bond acceptor/metal ligator center, hydrogen bond donor center, and aromatic center/ hydrophobic area. These features were consistent with the antiTB activity of the tested compounds.

|
Download:
|
| Figure 6. Heteryl subtstituted isatins nalidixic acid Schiff bases 7. | |
{kind=link}
According to the prediction of World Health Organization, at least one-third of the 34 million people with HIV infection worldwide are infected with MTB, and they are 21-34 times more likely to develop active TB than people without the HIV infection. As a result, TB is the leading cause of death for AIDS patients. Some FQs-isatin hybrids exhibited an inhibitory activity on HIV replication [19, 20]. Banerjee et al. [21] reported novel FQs-isatin hybrids 8, which could suppress HIV-replication and inhibit the MTB (Fig. 7) . Presence of various FQs at R guides activity against MTB H37Rv and HIV-1 (IIIB) cells in the following sequence: gatifloxacin > lomefloxacin > ciprofloxacin ≈ norfloxacin. The most active hybrid, 8c, displayed excellent activity against MTB H37Rv (MIC: 1.19 μmol/L), which was proved to effectively inhibit the replication of HIV-1 cells (EC50: 15.35μmol/L). In the cytotoxicity study on MT-4 cells, hybrid 8c showed 50% cell viability (CC50) at the concentration of 233.73 μmol/L, which was less cytotoxic than the references nevirapine (CC50: 156 μmol/L) and delaviridine (CC50: 87 μmol/L). Sriram et al. [22] also synthesized FQs-isatin lamivudine prodrugs 9 and evaluated their antimycobacterial activity against MTB H37Rv at a concentration of 6.25 μg/mL in BACTEC 12B medium using the BACTEC 460 radiometric system (Fig. 7) . The usefulness of the prodrugs of lamivudine should depend not only on the stability of the prodrug for its transport across the cell membrane, but also upon its intracellular reversion to the parent compound, especially in the virally infected cells. The half-lives (t1/2) of hydrolysis of the prodrugs were therefore determined. The data indicated that the various prodrugs of lamivudine were susceptible to hydrolysis with t1/2 in the range of 2-4 h. The in vitro result indicated that all the synthesized Mannich bases were found to be most active with 92%-100% inhibition while the parent lamivudine did not show any activity against MTB H37Rv at 6.25 μg/mL. The introduction of a fluorine group at the C-5 position of isatin enhanced its activity against HIV-1, whereas the introduction of 5-chloro group increased the toxicity to the CEM cell line. The most active compound (R1=F, R= norfloxacin) inhibited HIV-1 replication and showed 100% inhibition against MTB H37Rv in the preliminary screening. Therefore, these prodrugs would be beneficial for the effective treatment of HIV/AIDS.

|
Download:
|
| Figure 7. FQs-isatin hybrids 8 and 9 with anti-HIV-and anti-TB activities. | |
{kind=link}
2.2. Isatin-thiazole hybrids
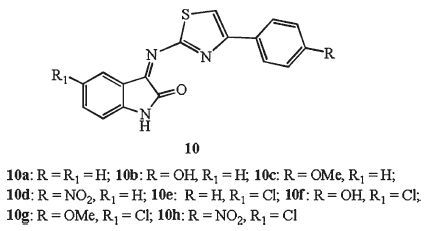
Joy and Mathew studied the antimycobacterial activity profile of various isatin imines incorporated with 4-substituted phenyl 2-amino thiazole 10 [23] (Fig. 8) . The result indicated that the presence of para hydroxyl and methoxyl group in the phenyl ring improves solubility and is a crucial factor for hydrogen bonding with receptor. The most active molecule was 10b, with a MIC of 6.25 μg/mL. The author believed that the combination of more electron donating and hydrogen bonding participating groups in the 4-and 6-position of phenyl and isatin system may generate more promising anti-TB candidates.

|
Download:
|
| Figure 8. Isatin-thiazole hybrids 10. | |
{kind=link}
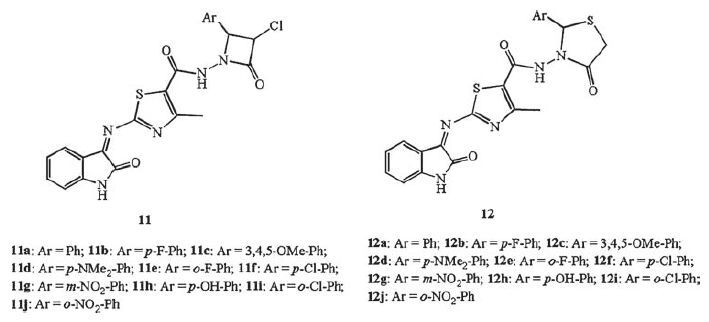
Two series of isatin-thiazole hybrids, 11 (azetidinone derivatives), and 12 (thiazolidinone derivatives), were synthesized and screened for their anti-TB activity [24] (Fig. 9) . The structure-activity relationship revealed that compounds having halogen were more potent than the others. Among of them, hybrids 11e, 11i and 12i were found to have promising antimycobacterial activity. Hybrid 11i emerged as the most promising candidate, with MIC of 0.39 μmol/L for MTB H37Rv.

|
Download:
|
| Figure 9. Isatin-thiazole azetidinone hybrids 11 and thiazolidinone hybrids 12. | |
{kind=link}
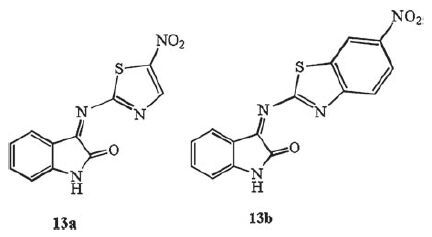
The N-position, C-3 and C-5 position modification of isatin by Jeankumar et al. was evaluated in vitro for their ability to inhibit MTB enzyme chorismate mutase (CM), and activity against MTB [25]. The isatin-thiazole hybrids 13a and 13b exhibited moderate activity against MTB CM (>100 μmol/L) and MTB (MIC: 77.16-91.24 μmol/L) (Fig. 10) .

|
Download:
|
| Figure 10. Isatin-thiazole hybrids 13 with nitro group. | |
{kind=link}
2.3. Tetrahydropyrimidine-isatin hybrids
The SAR study indicated that the substituent groups on the tetrahydropyrimidine ring were critical for activity, while incorporation of thiadiazole heterocycle would enhance the lipophilicity of the compounds, which was deemed important for the compounds to penetrate into TB cells [26]. Inspired by the biological properties of tetradihydropyrimidines and 5-substituted isatin, Akhaja and Raval [27] incorporated thiadiazoles and Schiff bases into isatin. Most of the tetrahydropyrimidine-isatin hybrids 14 displayed moderate activity. Hybrids 14f (MIC: 100 μg/mL) and 14l (MIC: 62.5 μg/mL) exhibited the highest activity against MTB H37Rv, which was comparable to Rifampicin (MIC: 40 μg/mL). Another series of tetrahydropyrimidine-isatin hybrids, 15, was further investigated [27]. The preliminary results indicated that compounds 15c, 15f, 15i and 15l exhibited the highest inhibition (99%) against MTB H37Rv at a constant concentration level (6.25 μg/mL). It was noted that compounds 15c and 15l displayed complete inhibition of MTB H37Rv (99%) at the MIC of 3.10-3.12 μg/mL. All of the remaining compounds showed moderate to good activity, with 74-99% inhibition at the concentration of 100-500 μg/mL. The SAR study indicated that the anti-TB activity decreased dramatically once 1, 3, 4-oxadiazole was replaced by 1, 3, 4-thiadiazole 16 [28] (Fig. 11) .

|
Download:
|
| Figure 11. Tetrahydropyrimidine-isatin hybrids 14-16. | |
{kind=link}
2.4. Thiolactone-isatin hybrids
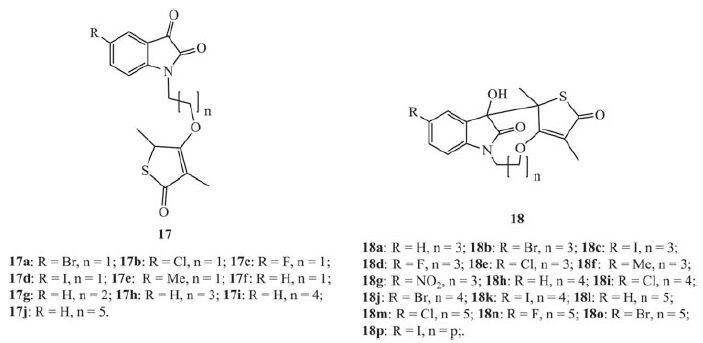
The key intermediate of the natural product antibiotic thiolactomycin (TLM) 17 is thiolactone 18, meanwhile the current study indicated that 4-O-alkylation of thiolactone 18 derivatives have exhibited potential activity against MTB [29]. Therefore, substituting at the 4-position of the thiolactone ring with isatin may yield promising anti-TB agents.
Hans et al. reported the antimycobacterial activity profile and in vitro cytotoxic evaluation of a series of thiolactone-isatin hybrids 17, as well as tetracyclic byproducts 18 [30]. According to the MABA assay, with the exception of 17b (MIC: 63.7 μmol/L), the MICs of all the targets were greater than the highest concentration tested (128 μmol/L). The low activity was also reflected by the BACTEC results, especially for the tetracycles, 18, which indicates the stimulation of mycobacterial growth (Fig. 12) . These results clearly suggest that the hybrids do not merit further investigation as antiTB agents.

|
Download:
|
| Figure 12. Thiolactone-isatin hybrids 17 and 18. | |
{kind=link}
2.5. Isoniazide-isatin hybrids
Isoniazid (INH), which is one of the most important first-line anti-TB agents, exerts its anti-TB activity by interfering with the synthesis of mycolic acid [31]. The main metabolic pathway for INH in humans is enzymatic acetylation of INH via N-acetyltransferase [31, 32], so proper modification of INH to block acetylation is one strategy to search a potent anti-TB agent with fewer side effects. Incorporating the pharmacophores of INH into isatin to develop novel INH-isatin hydrazides may lead to promising anti-TB agents.
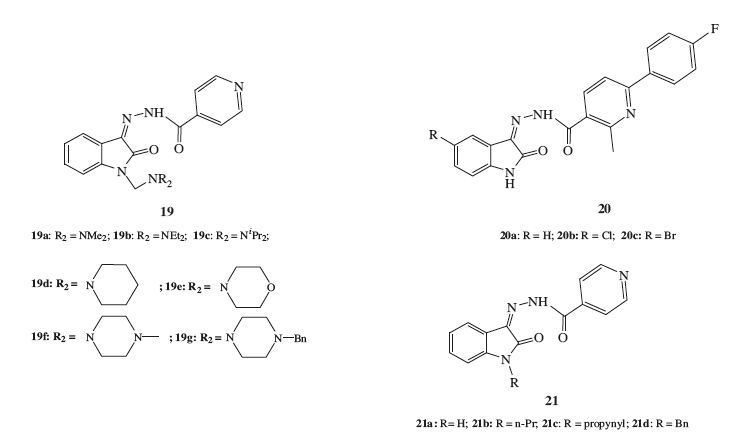
A series of INH-isatin hybrids 19 (Fig. 13) with increased lipophilicity (Clog P: 1.727-4.429) compared with INH (Clog P: -0.708) were screened for their activity against bovine MTB. Although all of the targets have the potential activity (3.5-4.5 μg/mL) against bovine MTB, these compounds were less active than the parent INH (1.5 μg/mL) [33].

|
Download:
|
| Figure 13. Isoniazide-isatin hybrids 19-21. | |
{kind=link}
The effect of substitution at the C-5 position of isatin moiety in hydrazides 20a-c (Fig. 13) was investigated by Eldehna et al. [31]. The results showed that 20a (Clog P: 3.54) , with an unsubstituted isatin moiety, displayed moderateanti-TB activity(MIC: 25 μg/mL). The introduction of a chlorine atom at the C-5 position (compound 20b, Clog P: 4.19) resulted in increased activity with MIC of 12.50 μg/mL. Incorporation of a bromine substituent (compound 20c, Clog P: 4.32) led to a remarkable increase of activity against MTB (MIC: 6.25 μg/mL). The order of activities of the isatin hydrazides increased in the order H < Cl < Br, which corresponds with the lipophilicity of the targets.
Aboul-Fadl et al. [6] performed the synthesis, in vitro anti-TB activity evaluation, and pharmacokinetic study of a series of 1-alkylisatin-INH hybrids 21 against three different strains: Bonin, human sensitive, and human resistand mycobacteria. The result revealed that hybrids 21a-d (Fig. 13) , with 1-unsubstituted, 1-propyl, 1-propynyl and 1-benzyl groups respectively, displayed equipotent growth inhibitory activity (MIC: 10 μmol/L) against the three tested strains as compared with INH. Compared with INH, these four hybrids exhibited potent activity against the human resistant strain. Pharmacokinetic study indicated that the rate and extent of absorption of the tested targets (21b and 21c) was significantly higher than that of INH (p < 0.05) . The relative bioavailabilities (FR%) were 183.15 and 443.25 for 21c and 21b, respectively, as compared to INH. These results further indicated potential of using this kind of hybrid for treatment of TB infections to overcome the resistance strains developed with INH.
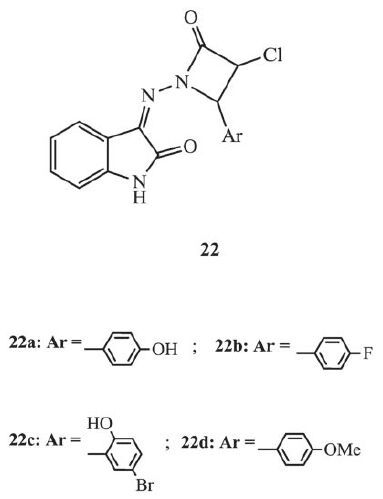
2.6. Azetidinone-isatin hybridsAzetidinone is a very useful pharmacophore. The antibacterial activity of some antibiotics, such as penicillins, cephalosporins, and carbapenems, is related to the 2-azetidinone ring in their skeletons. Moreover, besides typical antibacterial activity, anti-TB activity has also been reported in compounds in the presence of 2-azetidinone ring [34]. Four azetidinone-isatin hybrids 22 (Fig. 14) were selected for anti-TB evaluation using in-silico modeling by Arul and Sunisha [34]. All of the hybrids displayed the moderate anti-TB activity with 73%-83% inhibition at the concentration of 250 μg/mL, and further modification could improve their anti-TB activity.

|
Download:
|
| Figure 14. Azetidinone-isatin hybrids 22. | |
{kind=link}
2.7. Benzimidazole-isatin hybrids
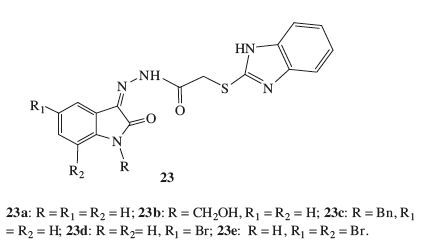
Introduction of benzimidazole ring is a promising strategy, which is an important pharmacophore for diverse pharmacological activities, including antibacterial and anti-virus activities. Therefore, hybridization of benzimidazoles. Benzimidazole-isatin hybrids 23 (Fig. 15) were synthesized and screened for their activity against MTB. However, all of the synthesized targets were less active than the reference RIF [35].

|
Download:
|
| Figure 15. Benzimidazole-isatin hybrids 23. | |
{kind=link}
2.8. 7-Chloroquinoline-isatin hybrids
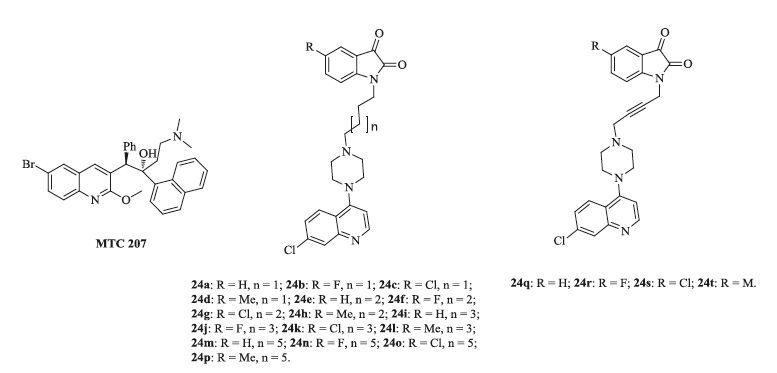
Diarylquinoline TMC 207 (Fig. 16) has not only shown potential activity against TB including MDR-TB, but also possessed a new mechanism of action based on interaction with the mycobacterial adenosine triphosphate (ATP) synthase [36, 37]. In addition, the quinoline derivatives such as 4-aminoquinoline and chloroquinoline, usually have low side effects and high efficacy, so proper modification of 4-amino-7-chloroquinoline may provide efficient anti-TB agents.

|
Download:
|
| Figure 16. Chemical structures of MTC 207 and 7-chloroquinoline-isatin hybrids 24. | |
{kind=link}
A series of twenty piperazine-tethered 7-chloroquinolineisatin hybrids 24 (Fig. 16) have been synthesized, and these new conjugates were evaluated for their anti-TB efficacy against MTB [37]. The result indicated that all of the hybrids are less active than that of INH (MIC: 0.18 μmol/L). The SAR study explicated that the presence of substituent at C-5 position of the isatin ring tends to influence the activity profile. In contrast, longer alkyl chain lengths adversely affect the anti-TB efficacy of the tested hybrids. It is noted that fluoro substituent is favorable for anti-TB efficacy, such as 24f and 24j (MIC: <10.12 μmol/L), which are >6 times more potent than cephalexin (MIC: 68.41 μmol/L). The Mannich adducts, on the other hand, proved to be inefficient in inhibiting the growth of MTB.
2.9. Thiacetazone-isatin hybridsIn recent years, thiacetazone derivatives have attracted attention due to their excellent activity against MTB, Mycobacterium avium, and other mycobacterial species [38-40]. The SAR study of isatin derivatives revealed that 5-halogenation, N-alkylation, and N-Mannich base, as well as the 3-thiosemicarbazone fragment, were critical to activity against various bacteria, fungi and viruses. Moreover, cyclization of isatins to 4-thiazoline, 4-thiazolidinone, and pyridazinoindole was an efficient way to increase their antimicrobial activity [10].
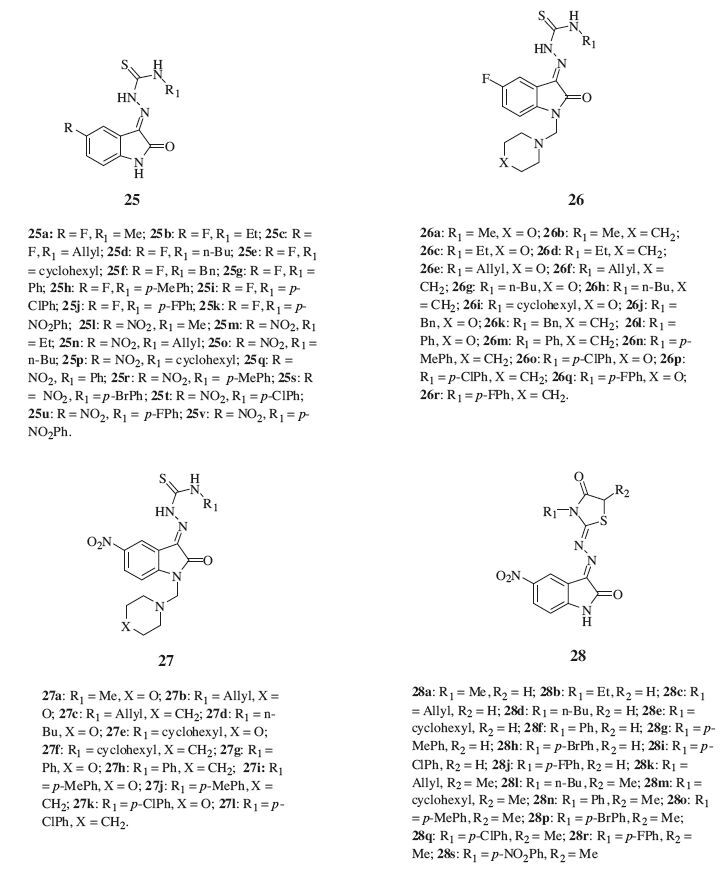
Based on the above considerations, four series of thiacetazoneisatin hybrids 25-28 (Fig. 17) were synthesized in order to obtain new and more potent anti-TB candidates [10, 41, 42]. Among the tested compounds, 5-nitro thiacetazone-isatin hybrids (24p, 24r, and 24s), and their 1-morpholinomethyl derivatives (26a, 26e, 26g, and 26i), exhibited significant inhibitory activity in the primary screen: the growth inhibition (GI) of virulent MTB H37 Rv was 90%-96% at 6.25 μg/mL. The SAR study revealed that halogenation in the C-5 position, elongation of the alkyl chain in the R1, replacement of the alkyl in the R1 with cyclohexyl or nonsubstituted phenyl, and the presence of substitution in the N-1 position were efficient methods in increasing anti-TB activity. 5-Nitro thiacetazone-isatin hybrids are generally more active than corresponding 5-fluoro analogs, and the presence of the morpholine ring in some 5-nitro thiacetazone-isatin hybrids also seems to have a significant impact on the resultant anti-TB activity. However, cyclized thiacetazone-isatin hybrids 27 showed the lowest activity (GI: 0-28% at 6.25 μg/mL) among the four series. In the light of these findings, a series of novel 5-methyl/ trifluoromethoxy thiacetazone-isatin hybrids were synthesized in order to obtain more potent and less toxic anti-TB candidates [43, 44]. In general, the author found that replacement of the alkyl in the R1 with cyclohexyl, phenyl, 4-fluorophenyl, 4-chlorophenyl, or 4-bromophenyl could increase the anti-TB activity. The increase of activity against MTB by alkyl chain elongation is related to lipophilicity, as confirmed by the log P value of these compounds.

|
Download:
|
| Figure 17. Thiacetazone-isatin hybrids 25-28. | |
{kind=link}
3. Conclusion
TB is one of the major life-threatening infectious diseases worldwide. The major issues in current TB treatment include the pathogen’s latency, the incidence of co-infection with HIV, poor patient compliance, and drug resistance issues caused by the emergence of MDR-TB and the recent advent of extensively drug resistant TB. Several promising anti-TB candidates have been found by three main approaches to developing novel anti-TB agents: (1) Expanding the antibacterial spectrum by applying existing drugs to the treatment of TB. (2) Searching for novel structures and mechanisms that have never been used to act against the TB organism. (3) Synthesizing new analogs or modifying existing drug compounds that can shorten and improve TB treatment. Hereinto, one of the most effective ways to produce novel anti-TB agent is to combine the pharmacophore moieties of different bioactive substances to give a new hybrid compound with improved affinity and efficacy compared to the parent drugs. Isatins are excellent reservoirs of bioactive substances, and the stability of the indole nucleus has inspired medicinal chemists to introduce many pharmacophore moieties into this nucleus and synthesize new potential anti-TB agents. Combining the pharmacophore moiety of isatin with different bioactive substances such as FQs, thiazole opens a door for the opportunities on the development of anti-TB agents. It is anticipated that in the near future some of the isatin hybrids will reach the clinical market for the treatment of TB.
| [1] | L.S. Feng, M.L. Liu, B. Wang, Synthesis and in vitro antimycobacterial activity of balofloxacin ethylene isatin derivatives. Eur. J. Med. Chem. 45 (2010) 3407–3412. DOI:10.1016/j.ejmech.2010.04.027 |
| [2] | T. Aboul-Fadl, F.A.S. Bin-Jubair, O. Aboul-Wafa, Schiff bases of indoline-2,3-dione (isatin) derivatives and nalidixic acid carbohydrazide, synthesis, antitubercular activity and pharmacophoric model building. Eur. J. Med. Chem. 45 (2010) 4578–4586. DOI:10.1016/j.ejmech.2010.07.020 |
| [3] | J. Crofton, P. Chaulet, D. Maher, Guidelines for the Management of Drugresistant Tuberculosis:WHO/TB/96-210(Rev. 1), World Health Organization, Geneva, 1997. |
| [4] | C. Viegas-Junior, A. Danuello, V. da Silva Bolzani, E.J. Barreiro, C.A.M. Fraga, Molecular hybridization:a useful tool in the design of new drug prototypes. Curr. Med. Chem. 14 (2007) 1829–1852. DOI:10.2174/092986707781058805 |
| [5] | A. González, J. Quirante, J. Nieto, Isatin derivatives, a novel class of transthyretin fibrillogenesis inhibitors. Bioorg. Med. Chem. Lett. 19 (2009) 5270–5273. DOI:10.1016/j.bmcl.2009.03.004 |
| [6] | T. Aboul-Fadl, F.A.H. Mohammed, E.A.S. Hassan, Synthesis, antitubercular activity and pharmacokinetic studies of some schiff bases derived from 1-alkylisatin and isonicotinic acid hydrazide (inh). Arch. Pharm. Res. 26 (2003) 778–784. DOI:10.1007/BF02980020 |
| [7] | S.K. Sridhar, M. Saravanan, A. Ramesh, Synthesis and antibacterial screening of hydrazones, Schiff and Mannich bases of isatin derivatives. Eur. J. Med. Chem. 36 (2001) 615–625. DOI:10.1016/S0223-5234(01)01255-7 |
| [8] | E. Piscopo, M.V. Diurno, R. Gogliadi, M. Cucciniello, G. Veneruso, Studies on heterocyclic compounds:indol-2,3-dione derivatives. VII. Variously substituted hydrazones with antimicrobial activity. Boll. Soc. Ital. Biol. Sper. 63 (1987) 827–832. |
| [9] | D. Sriram, T.R. Bal, P. Yogeeswari, Newer aminopyrimidinimino isatin analogues as non-nucleoside HIV-1 reverse transcriptase inhibitors for HIV and other opportunistic infections of AIDS:design, synthesis and biological evaluation. Farmaco 60 (2005) 377–384. DOI:10.1016/j.farmac.2005.03.005 |
| [10] | N. Karalı, A. Gürsoy, F. Kandemirli, Synthesis and structure-antituberculosis activity relationship of 1H-indole-2,3-dione derivatives. Bioorg. Med. Chem. 15 (2007) 5888–5904. DOI:10.1016/j.bmc.2007.05.063 |
| [11] | D. Sriram, A. Aubry, P. Yogeeswari, L.M. Fisher, Gatifloxacin derivatives:synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg. Med. Chem. Lett. 16 (2006) 2982–2985. DOI:10.1016/j.bmcl.2006.02.065 |
| [12] | D. Sriram, P. Yogeeswari, J.S. Basha, D.R. Radha, V. Nagaraja, Synthesis and antimycobacterial evaluation of various 7-substituted ciprofloxacin derivatives. Bioorg. Med. Chem. 13 (2005) 5774–5778. DOI:10.1016/j.bmc.2005.05.063 |
| [13] | S.N. Pandeya, D. Sriram, P. Yogeeswari, S. Ananthan, Antituberculous activity of norfloxacin mannich bases with isatin derivatives. Chemotherapy 47 (2001) 266–269. DOI:10.1159/000048533 |
| [14] | S. Talath, B.A. Bhongade, Synthesis, antimicrobial and anticancer studies of isatin derivatives of sparfloxacin. Am. PharmTech Res. 3 (2013) 570–581. |
| [15] | B.Y. Zhao, R. Pine, J. Domagala, K. Drlica, Fluoroquinolone action against clinical isolates of Mycobacterium tuberculosis:effects of a C-8 methoxyl group on survival in liquid media and in human macrophages. Antimicrob. Agents Chemother. 43 (1999) 661–666. |
| [16] | L.S. Feng, M.L. Liu, S. Zhang, Synthesis and in vitro antimycobacterial activity of 8-OCH3 ciprofloxacin methylene and ethylene isatin derivatives. Eur. J. Med. Chem. 46 (2011) 341–348. DOI:10.1016/j.ejmech.2010.11.023 |
| [17] | L.S. Feng, M.L. Liu, S. Wang, Synthesis and in vitro antimycobacterial activity of moxifloxacin methylene and ethylene isatin derivatives. Chem. Res. Chin. Univ. 28 (2012) 61–66. |
| [18] | Z.L. Wan, M.L. Liu, L.S. Feng, Synthesis and in vitro antimycobacterial activity of gatifloxacin ethylene isatin derivatives. Chin. J. Antibiot. 36 (2011) 37–43. |
| [19] | T.R. Bal, B. Anand, P. Yogeeswari, D. Sriram, Synthesis and evaluation of antiHIV activity of isatin β-thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 15 (2005) 4451–4455. DOI:10.1016/j.bmcl.2005.07.046 |
| [20] | D. Sriram, T.R. Bal, P. Yogeeswari, Aminopyrimidinimino isatin analogues:design of novel non-nucleoside HIV-1 reverse transcriptase inhibitors with broad-spectrum. J. Pharm. Pharm. Sci. 8 (2005) 565–577. |
| [21] | D. Banerjee, P. Yogeeswari, P. Bhat, Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co-infection. Eur. J. Med. Chem. 46 (2011) 106–121. DOI:10.1016/j.ejmech.2010.10.020 |
| [22] | D. Sriram, P. Yogeeswari, G. Gopal, Synthesis, anti-HIV and antitubercular activities of lamivudine prodrugs. Eur. J. Med. Chem. 40 (2005) 1373–1376. DOI:10.1016/j.ejmech.2005.07.006 |
| [23] | N. Joy, B. Mathew, Molecular hybridization and preclinical evaluation of imines from para-substituted 4-phenyl 2-amino thiazole incorporated with isatin analogues as antitubercular agents. Anti-Infect. Agents 13 (2015) 60–64. DOI:10.2174/2211352512666140905232639 |
| [24] | R.D. Dighe, S.S. Rohom, P.D. Dighe, M.R. Shiradkar, Condensed bridgehead nitrogen heterocyclic systems:synthesis and evaluation of isatinyl thiazole derivatives as anti-Mycobacterium tuberculosis agents and dTDP-rhamnose inhibitors. Der Pharma Chem. 3 (2011) 418–432. |
| [25] | V.U. Jeankumar, R. Alokam, J.P. Sridevi, Discovery and structure optimization of a series of isatin derivatives as Mycobacterium tuberculosis chorismate mutase inhibitors. Chem. Biol. Drug Des. 83 (2014) 498–506. DOI:10.1111/cbdd.2014.83.issue-4 |
| [26] | T.N. Akhaja, J.P. Raval, Design, synthesis, in vitro evaluation of tetrahydropyrimidine-isatin hybrids as potential antibacterial, antifungal and anti-tubercular agents. Chin. Chem. Lett. 23 (2012) 446–449. DOI:10.1016/j.cclet.2012.01.040 |
| [27] | T.N. Akhaja, J.P. Raval, Design, synthesis and in vitro evaluation of tetrahydropyrimidine-isatin hybrids as potential antitubercular and antimalarial agents. Chin. Chem. Lett. 23 (2012) 785–788. DOI:10.1016/j.cclet.2012.05.004 |
| [28] | T.N. Akhaja, J.P. Raval, 1,3-Dihydro-2H-indol-2-ones derivatives:design, synthesis, in vitro antibacterial, antifungal and antitubercular study, Eur. J. Med. Chem. 46(2011) 5573-5579. |
| [29] | A. Kamal, A.A. Shaik, R. Sinha, J.S. Yadav, S.K. Arora, Antitubercular agents. Part 2:new thiolactomycin analogues active against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 15 (2005) 1927–1929. DOI:10.1016/j.bmcl.2005.01.084 |
| [30] | R.H. Hans, I.J.F. Wiid, P.D. van Helden, Novel thiolactone-isatin hybrids as potential antimalarial and antitubercular agents. Bioorg. Med. Chem. Lett. 21 (2011) 2055–2058. DOI:10.1016/j.bmcl.2011.02.008 |
| [31] | W.M. Eldehna, M. Fares, M.M. Abdel-Aziz, H.A. Abdel-Aziz, Design, synthesis and antitubercular activity of certain nicotinic acid hydrazides. Molecules 20 (2015) 8800–8815. DOI:10.3390/molecules20058800 |
| [32] | J. Sandy, A. Mushtaq, A. Kawamura, The structure of arylamine Nacetyltransferase from Mycobacterium smegmatis-an enzyme which inactivates the anti-tubercular drug, isoniazid. J. Mol. Biol. 318 (2002) 1071–1083. DOI:10.1016/S0022-2836(02)00141-9 |
| [33] | M.A. Hussein, T. Aboul-Fadl, A. Hussein, Synthesis and antitubercular activity of some mannich bases derived from isatin isonicotinic acid hydrazone. Bull. Pharm. Sci.:Assiut Univ. 28 (2005) 131–136. |
| [34] | K. Arul, K.S. Sunisha, In-silico design, synthesis and in vitro anticancer and antitubercular activity of novel azetidinone containing isatin derivatives. Int. J. Pharm. Pharm. Sci. 16 (2014) 506–513. |
| [35] | S.K. Gupta, S.S. Pancholi, Synthesis and evaluation of antitubercular activity of some thiobenzimidazolyl derivatives. Der Pharma Chem. 3 (2011) 274–279. |
| [36] | A.H. Diacon, A. Pym, M. Grobush, The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 360 (2009) 2397–2405. DOI:10.1056/NEJMoa0808427 |
| [37] | R. Raj, C. Biot, S. Carrère-Kremer, L. Kremer, et al., 7-Chloroquinoline-isatin conjugates:antimalarial, antitubercular, and cytotoxic evaluation, Chem. Biol. Drug Des. 83(2014) 622-629. |
| [38] | M.T. Cocco, C. Congiu, V. Onnis, M.L. Pellerano, A. De Logu, Synthesis and antimycobacterial activity of new S-alkylisothiosemicarbazone derivatives. Bioorg. Med. Chem. 10 (2002) 501–506. DOI:10.1016/S0968-0896(01)00310-8 |
| [39] | L.E. Bermudez, R. Reynolds, P. Kolonoski, Thiosemicarbazole (thiacetazone-like) compound with activity against Mycobacterium avium in mice. Antimicrob. Agents Chemother. 47 (2003) 2685–2687. DOI:10.1128/AAC.47.8.2685-2687.2003 |
| [40] | A. De Logu, M. Saddi, V. Onnis, et al., In vitro antimycobacterial activity of newly synthesised S-alkylisothiosemicarbazone derivatives and synergistic interactions in combination with rifamycins against Mycobacterium avium, Int. J. Antimicrob. Agents 26(2005) 28-32. |
| [41] | N. Karalı, Synthesis and primary cytotoxicity evaluation of new 5-nitroindole-2,3-dione derivatives. Eur. J. Med. Chem. 37 (2002) 909–918. DOI:10.1016/S0223-5234(02)01416-2 |
| [42] | N. TerzioÐlu, N. Karalı, A. Gürsoy, Synthesis and primaryantiviral activity evaluation of 3-hydrazono-5-nitro-2-indolinone derivatives. ARKIVOC 1 (2006) 109–118. |
| [43] | Ö. Güzel, N. Karalı, A. Salman, Synthesis and antituberculosis activity of 5-methyl/trifluoromethoxy-1H-indole-2,3-dione-3-thiosemicarbazone derivatives. Bioorg. Med. Chem. 16 (2008) 8976–8987. DOI:10.1016/j.bmc.2008.08.050 |
| [44] | M. Shahlaei, A. Fassihi, A. Nezami, QSAR study of some 5-methyl/trifluoromethoxy-1H-indole-2,3-dione-3-thiosemicarbazone derivatives as anti-tubercular agents, Res. Pharm. Sci. 4(2009) 123-131. |