 2017, Vol. 28
2017, Vol. 28 



Alangium chinense (Lour.) Harms is a deciduous shrub belonging to family Alangiaceae and distributes mainly in China. The roots, flowers, and leaves of this plant have historically been applied as a traditional Chinese medicine for the treatment of rheumatoid arthritis, traumatic injury, fracture, and pain [1]. Alkaloids, terpenoids and phenolics have been previously reported from this plant [2-4]. As part of a program to study the bioactive substances from medicinal plants, an ethanolic extract of dried roots of A. chinense was investigated. Three new (1-3) and twenty-eight known phenolics (4-31) [5-25] were identified (the names of the known compounds were deposited in Supporting information), and their antiviral/antioxidant bioactivities were also evaluated.
2. Experimental 2.1. General experimentalOptical rotations were recorded on a JASCO P-2000 automatic digital polarimeter. UV spectra were measured on a JASCO V650 spectrophotometer. CD spectra were recorded on a JASCO J-815 spectropolarimeter. IR spectra were recorded on a Nicolet 5700 FT-IR spectrometer. NMR spectra were recorded on INOVA-500 and SX-600 spectrometers. ESI-MS spectra were measured on an Agilent 1100 Series LC/MSD ion trap mass spectrometer. HR-ESI-MS data were recorded on an Agilent Technologies 6250 Accurate-Mass Q-TOF LC/MS spectrometer. EIMS and HREIMS data were recorded on an AutoSpec Ultima-TOF MS spectrometer. Preparative HPLC was performed on a Shimadzu LC-6AD instrument with an SPD-10A detector, using a YMC-Pack ODS-A column (250 mm × 20 mm, 5 μm). Sephadex LH-20 (Amersham Pharmacia Biotech AB, Sweden), ODS (45-70 μm, Merck), macroporous adsorptive resins (XAD-D101, Tianjin Nankai Chemical Inc., China), polyamide resin (30-60 mesh, Jiangsu Linjiang Chemical Inc., China), and silica gel (200-300 mesh, Qingdao Marine Chemical Inc., China) were used for column chromatography (CC). TLC was conducted with glass precoated with silica gel GF254 (Qingdao Marine Chemical Inc., China).
2.2. Plant materialThe roots of A. chinense were collected from Guangxi Province, China in July 2009 and identified by Prof. Peng-Fei Tu (Peking University, School of Pharmaceutical Sciences). A voucher specimen (ID-S-2356) has been deposited in the Herbarium of the Department of Medicinal Plants, Institute of Materia Medica, Chinese Academy of Medical Sciences, China.
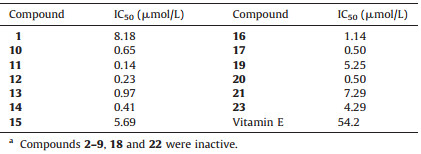
2.3. Extraction and isolationAir-dried, powdered roots of A. chinense (200 kg) were macerated for 12 h with 800 L of aqueous 95% EtOH and refluxed for 6 h (800 L × 3). After removal of the solvent under vacuum, the resultant residue (8 kg) was suspended in acidic H2O (100 L) and acidified to pH 2 with HCl to afford acidic H2O-soluble and acidic H2O-insoluble fractions. The acidic mixture was then filtered and partitioned with petroleum ether. The acidic H2O phase was basified to pH 10 with NaOH and then partitioned with CHCl3 to yield the CHCl3 extract (90 g). The alkaline H2O phase was then acidified to pH 7 with HCl and partitioned with n-BuOH to yield the n-BuOH extract (210 g). The crude CHCl3 extract (90 g) was fractionated using a basified silica gel column (pH 8-9, 200-300 mesh, 1.6 kg), eluting with petroleum ether containing increasing amounts of EtOAc (1:0, 50:1, 20:1, 10:1, 5:1, 1:1), and then eluted with CH2Cl2:MeOH (10:1-0:100) to afford eight fractions (A-H). Fraction F (7.2 g) was fractionated via an ODS column (45-70 μm, 400 g) by eluting with a gradient of MeOH (5%-100%) in H2O to yield six major fractions (F1-F5). Fraction F5 (1.2 g) was chromatographed over a Sephadex LH-20 column with CH2Cl2:MeOH (5:1) and was further purified by reversed-phase preparative HPLC (MeOH-H2O-TFA 40:60:0.03) to afford compound 1 (8 mg, Fig. 1). Fraction G (7.2 g) was fractionated via an ODS column (45-70 μm, 400 g) by eluting with a gradient of MeOH (5%-100%) in H2O to yield five major fractions (G1-G5). Fraction G3 (1.1 g) was chromatographed over a Sephadex LH-20 column with CH2Cl2:MeOH (5:1) and was further purified by reversed-phase preparative HPLC (MeOH-H2O-TFA 30:70:0.03) to afford compounds 3 (17 mg, Fig. 1).

|
Download:
|
| Figure 1. Structures of compounds 1-3 from A. chinense. | |
The crude n-BuOH extract (210 g) was fractionated using a macroporous adsorptive resins column (XAD-D101, 3.9 kg), eluting with D2O and then eluted with ethanol/D2O (95/5) to afford ethanol/D2O (95/5) fraction (135 g). The ethanol/D2O (95/5) fraction (135 g) was fractionated using a polyamide resins column (30-60 mesh, 2.0 kg), and eluted with D2O containing increasing amounts of ethanol (88:12, 75:25, 60:40, 5:95) to afford D2O fraction (86 g). The D2O fraction (86 g) was fractionated using a silica gel column (200-300 mesh, 1.5 kg), and eluted with petroleum ether containing increasing amounts of acetone (25:1, 10:1, 5:1, 3:1) and then with CH2Cl2:MeOH (20:1-0:100) to afford seven fractions (A-G). Fraction D (22.7 g) was fractionated via an ODS column (45-70 μm, 400 g) by eluting with a gradient of MeOH (5-100%) in H2O to yield five major fractions (D1-D5). Fraction D1 (3.4 g) was chromatographed over a Sephadex LH-20 column with MeOH:H2O (4:1) and was further purified by reversed-phase preparative HPLC (MeOH-H2O 22:78) to afford compound 2 (15 mg, Fig. 1).
Twenty-eight known compounds (4-31) were also isolated and identified from the roots of A. chinense, the detail were deposited in Supporting information.
(7R, 8R)-Threo-4, 7, 9, 90-tetrahydroxy-3, 5, 20-trimethoxy-8-O-40-neolignan (1): White amorphous powder; [a]D20 +55.2 (c 0.3, MeOH); UV (MeOH) λmax (log ε) 207 (4.73), 233 (4.08), 280 (3.57) nm; CD (MeOH) 234 (Δε -5.48), 253.5 (Δε +2.26), 313 (Δε +0.89) nm. IR (KBr) νmax 3399, 2940, 1678, 1613, 1513, 1462, 1426, 1327, 1262, 1220, 1118, 1032, 835, 802, 722 cm-1; 1H NMR (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) data, see Table 1; ESI-MS m/z 431 [M+Na]+, 407 [M-H]-; HR ESI-MS m/z 431.1676 [M+Na]+ (calcd. for C21H28O8Na, 431.1676).
|
|
Table 1 1H NMR and 13C NMR data of compounds 1-3 in CD3OD.a |

2-(Hydroxymethyl) phenol 1-O-β-D-glucopyranose-(1→6)-O-α-L-rhamno-pyranoside (2): White amorphous powder; [α]D20 +75.1 (c 0.6, MeOH); IR (KBr) υmax 3382, 2923, 1604, 1493, 1455, 1234, 1071, 761 cm-1; 1H NMR (500 MHz, CD3OD) and 13C NMR (125 MHz, CD3OD) data, see Table 1; ESIMS m/z 455 [M+Na]+, 431[M-H]-; HR ESI-MS m/z 455.1525 [M+Na]+ (calcd. for C19H28O11Na, 455.1524).
2-(Ethoxymethyl) phenol 1-O-β-D-glucopyranoside (3): White amorphous powder; [α]D20 +92.3 (c 0.8, MeOH); IR (KBr) υmax 3461, 3268, 2874, 1677, 1604, 1492, 1454, 1391, 1235, 1197, 1104, 1071, 893, 856, 754 cm-1; 1H NMR (500 MHz, CD3OD) and 13C NMR (125 MHz, CD3OD) data, see Table 1; ESI-MS m/z 337 [M+Na]+; HR ESI-MS m/z 337.1276 [M+Na]+ (calcd. for C15H22O7Na, 337.1258).
2.4. Acid hydrolysis of 2Compound 2 (3 mg) was dissolved in 2 mol/L HCl (aq) (2 mL) and heated at 90 ℃ for 10 h under constant stirring. After extraction with EtOAc (3× 2 mL), the aqueous layer was evaporated and cryodesiccated. Each residue was dissolved in dry pyridine (1 mL), and then L-cysteine methyl ester hydrochloride (4 mg) was added. Each mixture was stirred at 60 ℃ for 2 h, and then 0.4 mL of N-trimethylsilylimidazole was added, followed by heating to dryness at 60 ℃ for 2 h. Each dried reactant was partitioned between n-hexane and H2O (4 mL), and the n-hexane fraction was subjected to gas chromatography (GC) (column: DM-5, 0.25 mm × 30 m × 25 μm; detector: FID; temperature: 280 ℃; injector temperature: 260 ℃; carrier: N2 gas). The sugars from each reactant were identified by comparison of their retention times with those for authentic standards [tR: 24.77 min for D-glucose, 24.34 min for L-rhamnose].
2.5. Acid hydrolysis of 3Following the same method used for acid hydrolysis of 2, compound 3 (2 mg) was hydrolyzed to afford the sugar moieties. The sugars from each reactant were then identified by comparison of their retention times with those for authentic standards [tR: 19.84 min for D-glucose].
2.6. In vitro anti-Coxsackie virus B3 activity assayThe anti-Coxsackie virus B3 activity assay was determined using the same method as previously described [2]. Briefly, confluent Vero cells grown in 96-well microplates were infected with 100 median tissue culture infective doses (100TCID50) of Cox B3 virus. After 1 h of adsorption at 37 ℃, the monolayers were washed with phosphate buffered saline (PBS) and incubated at 37 ℃ in maintenance media (MEM plus 2% fetal bovine serum (FBS)) with or without different concentrations of test compounds. The viral cytopathic effect (CPE) was observed when the viral control group reached 4+, and the antiviral activity of the tested compounds was determined by Reed and Muench analyses.
2.7. Antioxidant assayThe antioxidant assays were conducted according to methods previously described [2]. Briefly, 1.0 mg of microsomal protein in 1 mL of 0.1 mol/L PBS buffer (pH 7.4) was incubated with 0.2 μmol/L cysteine and the test samples at 37 ℃ for 15 min. Lipid peroxidation was initiated by the addition of 0.05 mmol/L FeSO4. After incubation, 1 mL of 20% trichloroacetic acid was added to terminate the reaction. The mixture was centrifuged for 10 min at 3000 rpm. The supernatant was removed and reacted with 0.67% TBA for 10 min at 100 ℃. After cooling, the MDA (malondialdehyde, a compound produced during microsomal lipid peroxidation induced by Fe2+-cysteine, was detected using the thiobarbituric acid (TBA) method) was quantified by UV/vis (absorbance at 532 nm), from which the inhibition rate (IR) was calculated as IR [%]=100% -At/(Ap -Ac) × 100, where Ap, At, and Ac refer to the absorbance of Fe2+-cysteine, test compound, and control (solvent only), respectively. Vitamin E was selected as the positive control.
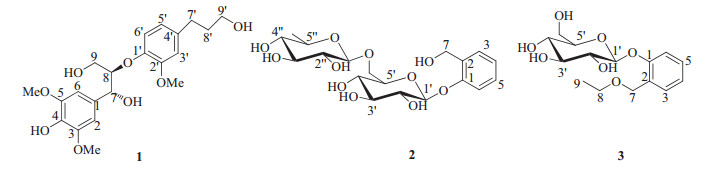
3. Results and discussionCompound 1, a white amorphous powder, displayed a molecular formula of C21H28O8 with 8 degrees of unsaturation, as established by HR-ESI-MS (m/z 431.1676 [M+Na]+, calcd. 431.1676). The IR spectrum showed the absorptions of hydroxyl group (3399 cm-1) and aromatic ring (1513 and 1462 cm-1). The NMR data of 1 (Table 1) indicated the presence of a symmetric 1, 3, 4, 5-tetrasubstituted phenyl, an 10, 20, 40-trisubstituted phenyl, three methoxyls, four methylenes (two oxygenated), and seven methine (two oxygenated) groups. Analysis of the 1D and 2D NMR data (including 1H-1H COSY, HSQC, HMBC spectra) allowed for the establishment of two C6-C3 units (A: C1-C9 and B: C1'-C9') of lignan in 1. The HMBC correlations (Fig. 2) from H-2/6 to C-7/C-4, from H-7 to C-2/6/9 and from H-9 to C-7, from H-8 to C-1, from OMe-3 to C-3 and from OMe-5 to C-5, together with their chemical shifts, led to the determination of unit C1-C9. Similarly, based on the HMBC correlations from H-3' to C-1'/C-5'/C-7', from H-5' to C-1'/C-3'/C-7', from H-6' to C-2'/C4', from H-7' to C-3'/C-5', from H-8' to C-4', from H-9' to C-7', from OMe-2' to C-2', the unit C1'-C9' was determined. The HMBC correlation from H-8 to C-1', together with the chemical shifts of C-8 and C-1', suggested that units A and B were connected through C8-O-C1' bond to form the planar structure, 4, 7, 9, 9'-tetrahydroxy-3, 5, 2'-trimethoxy-8-O-4'-neolignan.

|
Download:
|
| Figure 2. Key 1H-1H COSY (-) and HMBC (H→C) correlations of 1-3. | |
In the 1H NMR spectrum, the coupling constant (J7, 8=5.4 Hz) indicated the 7, 8-threo relative configuration [26]. A negative Cotton effect at 234 nm (Δε -5.48) in the CD spectrum suggested the 8R configuration for 1 [27-31]. Based on the relative configuration assigned above, the absolute configuration was determined to be 7R and 8R. Thus, 1 was characterized as shown in Fig. 1 and named to be (7R, 8R)-threo-4, 7, 9, 9'-tetrahydroxy-3, 5, 2'-trimethoxy-8-O-4'-neolignan.
Compound 2 was obtained as white amorphous powder, and its molecular formula was established as C19H28O11 by HR-ESIMS at m/z 455.1525 [M+Na]+ (calcd. for 455.1524), with 6 degrees of unsaturation. The IR spectrum showed the absorptions of hydroxyl and aromatic groups at 3382 and 1493 cm-1, respectively. The 13C NMR and DEPT spectra of 2 showed the presence of 19 carbon signals, including one methyl, two methylenes, fourteen methines, and two quaternary carbons in the structure. One 1, 2-disubstituted phenyl unit was assigned based on the chemical shifts and splitting pattern of four aromatic protons at dH 6.97-7.27 in the 1H NMR of 2 (Table 1), which were confirmed by the six 13C resonances at δC 117-157. The HMBC correlations (Fig. 2) from H-7 to C-1 (δC 157.2)/C-2 (δC 132.5)/C-3 (δC 130.2) demonstrated that one methylene group C-7 (δC 61.2) was attached at the C-2 to form the 2-(hydroxymethyl) phenol aglycone. The 1H NMR data displayed the signals of two anomeric protons at δH 4.78 (1H, d, 8.4 Hz) and 4.64 (1H, d, 1.35 Hz), indicating the existence of two mono-sugar moieties. Analysis of the 1H-1H COSY, HSQC, and acidic hydrolysis experiments allowed for the establishment of the two sugar moieties to be rhamnopyranosyl (C1"-C6") and glucopyranosyl (C1'-C6'), respectively. The HMBC correlation (Fig. 2) from H-1' to C-1 revealed that the glucopyranosyl moiety was connected at C-1 of the 2-(hydroxymethyl) phenol aglycone. Based on the HMBC correlations between H-6' and C-1" and between H-1" to C-6', the rhamnopyranosyl was determined to be linked at C-6' of the glucopyranosyl moiety. Thus, the planar structure of 2 was established to be 2-(hydroxymethyl) phenol 1-O-glucopyranose-(1→6)-rhamnopyranoside.
The splitting patterns of the anomeric proton H-1' (d, J=8.4 Hz) indicated that the glucopyranose was in β-configuration. The absolute configuration of β-glucopyranose was then confirmed to be D by acid hydrolysis and GC analysis. The relative configuration ofH-1" of rhamnopyranoside could not be assigned by the coupling constant of the anomeric proton. Therefore, the rhamnopyranoside was unambiguously determined to be α-L-rhamnopyranoside by acid hydrolysis and GC analysis. Thus, the structure of 2 was characterized to be 2-(hydroxymethyl) phenol 1-O-β-D-glucopyranose-(1→6)-O-α-L-rhamnopyranoside.
Compound 3 was obtained as white amorphous powder, and its molecular formula was established as C15H22O7 based on the HRESIMS at m/z 337.1276 [M+Na]+ (calcd. for 337.1258), with 5 degrees of unsaturation. Comparison the NMR data of 3 with 2 (Table 1) revealed that 3 was an analog of 2, a glucoside of 2-(hydroxymethyl) phenol, except for the absence of rhamnopyranosyl and the presence of one ethanol group in 3. Acid hydrolysis and GC experiment unambiguously confirmed the presence of β-Dglucose in the structure. The HMBC correlations (Fig. 2) from H-10 (anomeric proton of glucosyl) to C-1 (δC 157.4), from H-7 to C-1 (δC 157.4)/C-3 (δC 130.7)/C-8 (δC 67.0), and from H-8 to C-7 established that the glucopyranosyl moiety was located at C-1 and the ethoxymethyl group was located at C-2 of the phenyl. The resonance of the anomeric proton at δH 4.82 (1H, d, J=7.5 Hz, H-1') indicated a β-glycosidic linkage. Thus, the structure of 3 was characterized as shown and named to be 2-(ethoxymethyl) phenol 1-O-β-D-glucopyranoside.
Twenty-eight known phenolics (4-31) were identified based on their spectroscopic profiles (NMR, UV, MS, andCD) andcomparison to published data.
Two different assays (in vitro anti-Coxsackie virus B3 activity assay, and antioxidant assay) were carried out to evaluate the bioactivities of 1-31. Compounds 2-9, 18, and 22 showed no activity in the assay. Compound 11 exhibited antiviral activities against Coxsackie virus B3 with IC50 values of 16.89 mmol/L. Compounds 1, 10-17, 19-21, and 23 showed strong antioxidant activities against Fe2+-cysteine induced rat liver microsomal lipid peroxidation, with IC50 values of 0.14-8.18 mmol/L (Table 2). Among them, compound 11 inhibited the strongest antioxidant activity with the IC50 values of 0.14 μmol/L. In the same assay, the IC50 of the positive control, vitamin E, was 54.2 μmol/L, indicated that compounds 1, 10-17, 19-21, and 23 showed much strong bioactivity than vitamin E and indicated that these compounds had potential health benefit.
|
|
Table 2 Antioxidant activity of compounds 1, 10-17, 19-21, and 23.a |
{kind=link}
{kind=link}
4. Conclusion
In summary, three new phenolics (1-3) and twenty-eight known compounds (4-31) were isolated from an ethanolic extract of roots of A. chinense. Compound 11 exhibited antiviral activity against Coxsackie virus B3 with IC50 values of 16.89 μmol/L. Compounds 1, 10-17, 19-21, and 23 showed strong antioxidant activities against Fe2+-cysteine-induced rat liver microsomal lipid peroxidation, with IC50 values of 0.14-8.18 μmol/L. This investigation could shed new light on the further understanding of the bioactive chemical constituents of A. chinense.
AcknowledgmentsThis project was supported by the National Science and Technology Project of China (No. 2009ZX09311-004) and the National Natural Science Foundation of China (No. 201072234). We are grateful to the Department of Instrumental Analysis, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, for measuring the IR, UV, NMR, and MS spectra.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.05.012.
| [1] | Chiang Su New Medical College, Dictionary of Chinese Crude Drugs, Shanghai Scientific Technologic Publisher, Shanghai, 1978, pp. 24-26. |
| [2] | Y. Zhang, Y.B. Liu, Y. Li, Sesquiterpenes and alkaloids from the roots of Alangium chinense. J. Nat. Prod. 76 (2013) 1058–1063. DOI:10.1021/np4000747 |
| [3] | A. Itoh, T. Tanahashi, N. Nagakura, Glycosides of benzyl and salicylalcohols from Alangium chinense. Chem. Pharm. Bull. 49 (2001) 1343–1345. DOI:10.1248/cpb.49.1343 |
| [4] | A. Itoh, T. Tanahashi, I. Sanae, Five phenolic glycosides from Alangium chinense. J. Nat. Prod. 63 (2000) 95–98. DOI:10.1021/np990391z |
| [5] | F. Abe, T. Yamauchi, 9ǁ-Hydroxypinoresinol, 9a-hydroxymedioresinol and related lignans from Allamanda neriifolia. Phytochemistry 27 (1988) 575–577. DOI:10.1016/0031-9422(88)83144-3 |
| [6] | H. Otsuka, M. Takeuchi, S. Inoshiri, Phenolic compounds from Coix lachrymajobi var. Ma-yuen. Phytochemistry 28 (1989) 883–886. DOI:10.1016/0031-9422(89)80136-0 |
| [7] | L. Xiong, C.G. Zhu, J.G. Shi, Lignans and neolignans from Sinocalamus affinis and their absolute configurations. J. Nat. Prod. 74 (2011) 1188–1200. DOI:10.1021/np200117y |
| [8] | S. Ferreira Fonseca, J. De Paiva Campello, L.E.S. Barata, 13C NMR spectral analysis of lignans from Araucaria angustifolia. Phytochemistry 17 (1978) 499–502. DOI:10.1016/S0031-9422(00)89347-4 |
| [9] | F. Li, X.W. Yang, Three new neolignans from the aril of Myristica fragrans. Helv. Chim. Acta 90 (2007) 1491–1496. DOI:10.1002/(ISSN)1522-2675 |
| [10] | M. Mitsuo, K. Hiroyuki, K. Hiromu, Microbial oxidation of (+)-epimagnolin a by Aspergillus niger. Phytochemistry 35 (1994) 1191–1193. DOI:10.1016/S0031-9422(00)94820-9 |
| [11] | J.W. Wang, J.Y. Liang, L. Li, Chemical constituents from Gnetum parvifolium. Chin. J. Nat. Med. 4 (2006) 432–434. |
| [12] | F.R. Chang, Y.C. Chao, C.M. Teng, Y.C. Wu, Chemical constituents from Cassytha filiformis Ⅱ. J. Nat. Prod. 61 (1998) 863–866. DOI:10.1021/np970348g |
| [13] | S. Michel, F. Tillequin, M. Koch, Alcaloïdes des Écorces de tiges de Strychnos dinklagei. J. Nat. Prod. 45 (1982) 489–494. DOI:10.1021/np50022a024 |
| [14] | L.X. Zhou, Y. Ding, Studies on chemical constituents of Ligustrum obtusifolium Sieb. et Zucc. Chin. J. Chin. Mater. Med. 25 (2000) 541–543. |
| [15] | S. Nishibe, H. Tsukamoto, S. Hisada, Effects of O-methylation and O-glucosylation on carbon-13 nuclear magnetic resonance chemical shifts of matairesinol, (+)-pinoresinol and (+)-epipinoresinol. Chem. Pharm. Bull. 32 (1984) 4653–4657. DOI:10.1248/cpb.32.4653 |
| [16] | A. Jutiviboonsuk, H.J. Zhang, G.T. Tan, Bioactive constituents from roots of Bursera tonkinensis. Phytochemistry 66 (2005) 2745–2751. DOI:10.1016/j.phytochem.2005.09.025 |
| [17] | K. Yoshinari, N. Shimazaki, Y. Sashida, Flavanone xyloside and lignans from Prunus jamasakura bark. Phytochemistry 29 (1990) 1675–1678. DOI:10.1016/0031-9422(90)80144-6 |
| [18] | Y. Takeda, C. Mima, T. Masuda, Glochidioboside, a glucoside of (7S, 8R)-dihydrodehydrodiconiferyl alcohol from leaves of Glochidion obovatum. Phytochemistry 49 (1998) 2137–2139. DOI:10.1016/S0031-9422(98)00362-8 |
| [19] | F. Abe, T. Yamauchi, Lignans from Trachelospermum asiaticum (Tracheolospermum. Ⅱ). Chem. Pharm. Bull. 34 (1986) 4340–4345. DOI:10.1248/cpb.34.4340 |
| [20] | H. Achenbach, M. Loewel, R. Waibel, New lignan glycosides from Stemmadenia minima. Planta Med. 58 (1992) 270–272. DOI:10.1055/s-2006-961451 |
| [21] | L.Z. Lin, C.Q. Song, R.S. Xu, Chemical constituents of the anticancer plant Camptotheca acuminata Decne. ó. Ellagic acids from the fruits of Camptotheca acuminata Decne. Acta Chim. Sin. 37 (1979) 207–214. |
| [22] | S. De Rosa, A. De Giulio, G. Tommonaro, Aliphatic and aromatic glycosides from the cell cultures of Lycopersicon esculentum. Phytochemistry 42 (1996) 1031–1034. DOI:10.1016/0031-9422(96)00083-0 |
| [23] | H. Kijima, T. Ide, H. Otsuka, Water-soluble phenolic glycosides from leaves of Alangium premnifolium. Phytochemistry 44 (1997) 1551–1557. DOI:10.1016/S0031-9422(96)00760-1 |
| [24] | S. Lin, S.J. Wang, M.T. Liu, Glycosides from the stem bark of Fraxinus sieboldiana. J. Nat. Prod. 70 (2007) 817–823. DOI:10.1021/np0700467 |
| [25] | C. Zdero, F. Bohlmann, R.M. King, Diterpene glycosides and other constituents from Argentinian baccharis species. Phytochemistry 25 (1986) 2841–2855. DOI:10.1016/S0031-9422(00)83754-1 |
| [26] | P.G.M. Wuts, S.S. Bigelow, Gamma-alkoxyallylboronates as useful reagents for preparation of differentially protected diol derivatives. J. Org. Chem. 47 (1982) 2498–2500. DOI:10.1021/jo00133a059 |
| [27] | N. Matsuda, M. Kikuchi, Studies on the constituents of Lonicera species. X. Neolignan glycosides from the leaves of Lonicera gracilipes var. glandulosa MAXIM. Chem. Pharm. Bull. 44 (1996) 1676–1679. DOI:10.1248/cpb.44.1676 |
| [28] | A. Arnoldi, L. Merlini, Asymmetric synthesis of 3-methyl-2-phenyl-1, 4-benzodioxanes. Absolute configuration of the neolignans eusiderin and eusiderin C and D, J. Chem. Soc. Perkin Trans. 1 (1985) 2555–2557. |
| [29] | S.G. Liao, Y. Wu, J.M. Yue, Lignans from Wikstroemia hainanensis. Helv. Chim. Acta 89 (2006) 73–80. DOI:10.1002/(ISSN)1522-2675 |
| [30] |
(a) M.D. Greca, A. Molinaro, P. Monaco, et al., Lignans from Arum italicum, Phytochemistry 35(1994) 777-779; (b) C.H. Huo, H. Liang, Y.Y. Zhao, et al., Neolignan glycosides from Symplocos caudata, Phytochemistry 69(2008) 788-795. |
| [31] |
(a) M.L. Gan, Y.L. Zhang, S. Lin, et al., Glycosides from the root of Iodes cirrhosa, J. Nat. Prod. 71(2008) 647-654; (b) K.H. Kim, E. Moon, S.Y. Kim, et al., Lignans from the tuber-barks of Colocasia antiquorum var. esculenta and their antimelanogenic activity, J. Agric. Food Chem. 58(2010) 4779-4785; (c) L. Xiong, C.G. Zhu, Y.R. Li, et al., Lignans and neolignans from Sinocalamus affinis and their absolute configurations, J. Nat. Prod. 74(2011) 1188-1200; (d) Y.R. Li, W. Cheng, C.G. Zhu, et al., Bioactive neolignans and lignans from the bark of Machilus robusta, J. Nat. Prod. 74(2011) 1444-1452. |