 2016, Vol. 27
2016, Vol. 27 


 , Xiao-Mei Ling, Li-He Zhang, Zhen-Jun Yang
, Xiao-Mei Ling, Li-He Zhang, Zhen-Jun Yang
As an idiomatical class of anti-viral nucleosides, 6-cyclopropylamino guanosine analogs have been extensively investigated, leading to the discovery of Abacavir (5, ABC), the prodrug of carbovir (4, CBV). It is an important clinical therapeutic agent against HIV-1 targeting HIV reverse transcriptase [1, 2]. The bioconversion of ABC (with 5′-3H labeling) in CEM cells was investigated with HPLC as described in Fig. S1 in Supporting information) [3, 4], in which the conversion route of “ABC to ABC monophosphate (ABC-MP) to CBV-MP to CBV triphosphate (CBVTP)” was proposed to be the dominant pathway. Their work did not rule out the path of “ABC to aminoCBV to aminoCBV-MP to CBV-MP to CBV-TP” as an alternative metabolic pathway. An enzyme named 6-methyl AMP aminohydrolase, was shown to be responsible for the deamination of ABC-MP to CBV-MP. But the enzyme did not catalyze the deamination of adenosine and 2′- deoxyadenosine or their corresponding N6-substituted derivatives [5], due to the detection limit of the analytical method, the alternative deamination pathway of ABC remains a hypothesis.
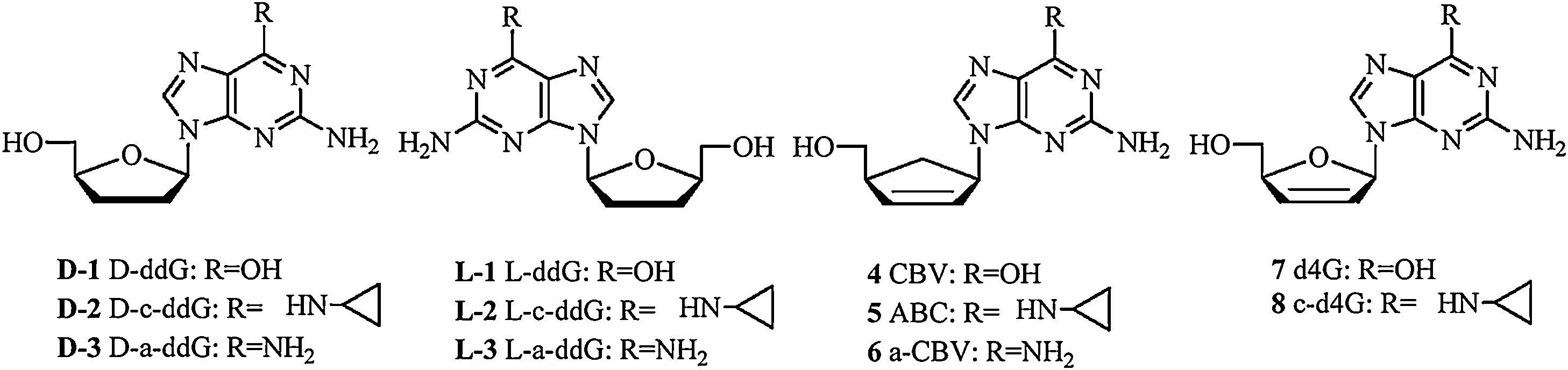
Most of the 2′, 3′-dideoxynucleosides (ddN) and 2′, 3′-didehydro- 2′, 3′-dideoxynucleosides (d4N) exhibit potent anti-viral activities as viral DNA polymerase inhibitors [6-8]. But 2′, 3′-didehydro- 2′, 3′-dideoxyguanosine (7, d4G) and 2′, 3′-dideoxyguanosine (D-1, ddG) showed less activity because of their solution instability and high polarity [9, 10]. 6-Cyclopropylamino-d4G (8, c-d4G) was synthesized as a prodrug of d4G, which showed to possess high stability and potent anti-HIV activity, it underwent similar metabolism in CEM and PBM cells as ABC [8]. L-Nucleosides are the enantiomer of natural D-configuration nucleosides, they were less toxic since their triphosphate active form can hardly be recognized by human DNA polymerases. Over the last 20 years, researchers have successfully developed two anti-viral drug Lamivudine and Telbivudine approved by FDA. To further exploit ddG/d4G prodrug as anti-viral drug, a series of 6-amino modified D-/L-ddG/d4G were synthesized in our previous work, D-form structures including 6-cyclopropylamino-ddG (D-2, D-c-ddG) displayed benign anti-HIV activities while their L-isomer were inactive [11, 12].
In this work, we developed an optimal capillary electrophoresis (CE) method without radiolabeling to analyze purine metabolites. It is efficient, rapid, and economic compared with traditional HPLC method and can be used as an alternative method for the analysis of nucleoside metabolites [13]. The metabolites of a series of guanosine analogs including D/L-(2) and 5 in mouse liver homogenate and microsome were analyzed (Fig. 1) through CE. Catalyzed deamination by ADA was also investigated. The products were identified based on migration times of standards and some of them were further confirmed by MS.

|
Download:
|
| Figure 1. Structures of guanosine analogs. | |
2. Experimental 2.1. Materials and instrumentation
All chemical reagents were of analytical grade unless otherwise indicated. PBS (pH 7.2-7.4) was from Sinopharm Chemical Reagent Beijing Co., Ltd. (Beijing, China). ADA was purchased from Beijing Dongge Biological Technology Co., Ltd (Beijing, China). Mouse liver microsome (20 mg/mL) and NADPH Regenerating System were obtained from iPhase Bioscience (Beijing, China). Pentostatin was purchased from Chemsky International Co., Ltd. (Shanghai, China). All nucleoside analogs were synthesized in our laboratory. CE analysis was performed on Beckman Proteomelab PA800 system (Beckman Coulter, Fullerton, CA, USA) equipped with an UV detector and 32 Karat software (Beckman). Capillary was purchased from Yongnian Optical Fiber Factory (Hebei province, China); LC-MS was carried out on the Shimadzu HPLC-Ion Trap- Time of Flight (LCMS-IT-TOF) mass spectrometer system.
2.2. Preparation of mouse liver homogenateThe mouse was sacrificed by cervical dislocation. Overnight fasting before sacrifice was used to reduce the glycogen content of the liver. The liver was removed rapidly and immersed in ice-cold saline. Ice-cold phosphate buffered saline (0.1 mol/L, 1 g/1 mL) was added. The liver was homogenized with an ultrasonic cell disruption system (200 W, 3 s ultrasonic time with 9 s interval, 9 times, ice-bath). Homogenate was centrifuged for 10 min at 12, 500 - g at 4 ℃ and the supernatant was carefully transferred into ultracentrifuge tubes for storage at -80 ℃.
2.3. Mouse liver homogenate samplesThe nucleoside stock solution was mixed with liver homogenate (50%) at the ratio of 1/1 (v/v) and incubated at 37 ℃. The mixture was deactivated at 94 ℃ for 10 min and centrifuged at 12500 g at 4 ℃ for 10 min. The supernatant was removed and analyzed by CE or LC-MS.
2.4. ADA metabolism samplesADA solution and nucleoside stock solution were mixed with equal volume on ice bath. The mixture was incubated at 37 ℃ for 3 h and heated at 85 ℃ for 10 min to terminate the reaction. The solution was centrifuged at 12500 g at 4 ℃ for 10 min. The supernatant was removed and analyzed by CE.
2.5. Mouse liver microsome samplesTo a mixture of solution A (10 mL) and solution B (2 mL) of NADPH Regenerating System was added nucleoside stock solution (5 mmol/L, 19 mL) on ice-bath. The mixture was preheated at 37 ℃ for 5 min and then liver microsome (20 mL) was added. After shaking mildly for a while, the mixture was incubated at 37 ℃ for 4 h and terminated at 85 ℃ for 10 min. The solution was centrifuged at 12, 500 - g at 4 ℃ for 10 min. The supernatant was removed and analyzed by CE. In some experiments, pentostatin solution was added into liver microsome incubation solution of D-c-ddG and the final incubation concentration was 0.5 mmol/L. The mixture was incubated at 37 ℃ for different times and terminated at 85 ℃ for 10 min.
2.6. CE analysisThe MECC mode of CE can be used for neutral substance separation and thus was applied in this work, analysis was performed with uncoated fused silica capillary of 75 mm inside diameter (i.d.) and 40 cm effective separation length (375 mm o.d. - 40.2 cm length) using micellar electrokinetic capillary chromatography (MECC) as the separation mode. Before use, new capillaries were rinsed with NaOH solution (0.1 mol/L) for 20 min and deionized water for 5 min. Separated bands were detected by UV absorption at 254 nm. The other analysis conditions are shown in Table S1 in Supporting information.
2.7. LC-MS analysisLC-MS analysis of D-c-ddG in liver homogenate: The sample solution aforementioned was diluted with acetonitrile (two times of sample volume) and centrifuged at 4 ℃ for 10 min (12, 500 - g). The supernatant was analyzed using reverse phase HPLC on a Waters Symmetry1C-18 column (5 mm, 100Å). Elution of nucleosides were accomplished with a solution of acetonitrile/H2O 11/89 (v/v) at a flow rate of 0.7 mL/min. MS was carried out under the following condition: split 0.21 mL/min; Injection volume: 2 mL; CDL TEM: 200 ℃; Heat Block Tem: 200 ℃; Nebulizing Gas: 1.5 L/min; Drying Gas: 100 kPa; Detector: 1.6 kV.
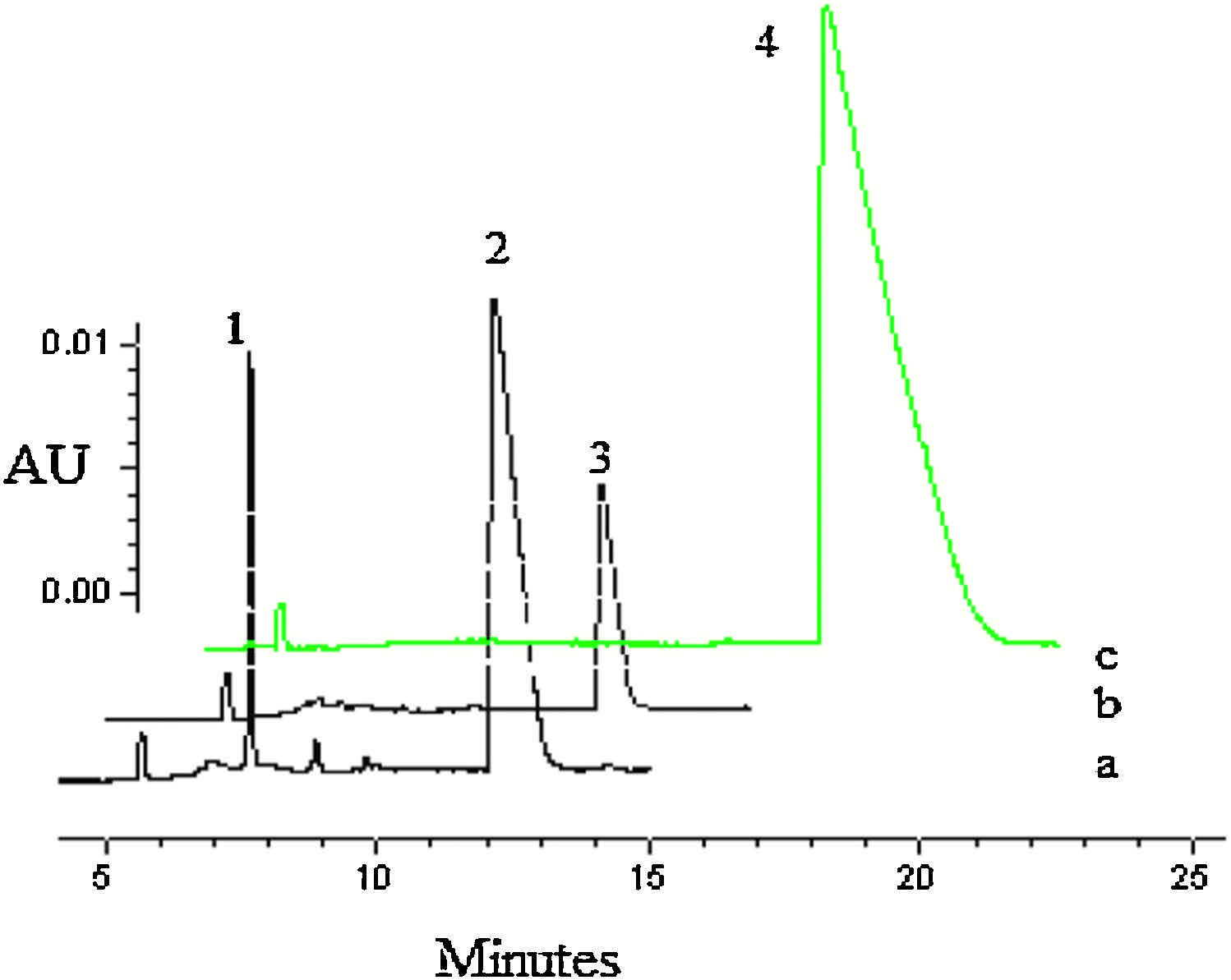
3. Results and discussion 3.1. Metabolism in mouse liver homogenateSmall portion of D-c-ddG was converted to D-a-ddG after incubation with mouse liver homogenate for 4 h (Fig. 2, curve b). Formation of D-a-ddG could be confirmed by LC-MS, in which the molecular weight of peak 1 (251.1237, [M+H]+) was in agreement with that of D-a-ddG (251.1237, [M+H]+) (Fig. S2 in Supporting information). These results suggested that D-c-ddG was firstly converted to D-a-ddG by oxidases in liver homogenate and then deaminated to D-ddG. The L-form counterparts were stable in mouse liver homogenate when treated under the same conditions.

|
Download:
|
| Figure 2. CE analysis of deamination metabolism of D/L-c-ddG incubated in mouse liver homogenate. a: liver homogenate as blank control; b: D-c-ddG, 4 h; c: L-c-ddG, 4 h; Peak 1: D-a-ddG; Peak 2: D-ddG; Peak 3: D-c-ddG; Peak 4: L-c-ddG. | |
For the other guanosine analogs, after incubated in liver homogenate for 4 h, only a small quantity of ABC was converted to 6-amino-CBV (a-CBV) and CBV after 4 h (Table 1).
|
|
Table 1 Bioconversion of 6-amino or 6-cyclopropylamino guanosine analogs. |
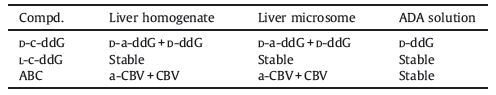
{kind=link}
{kind=link}
3.2. Oxidation in mouse liver microsome
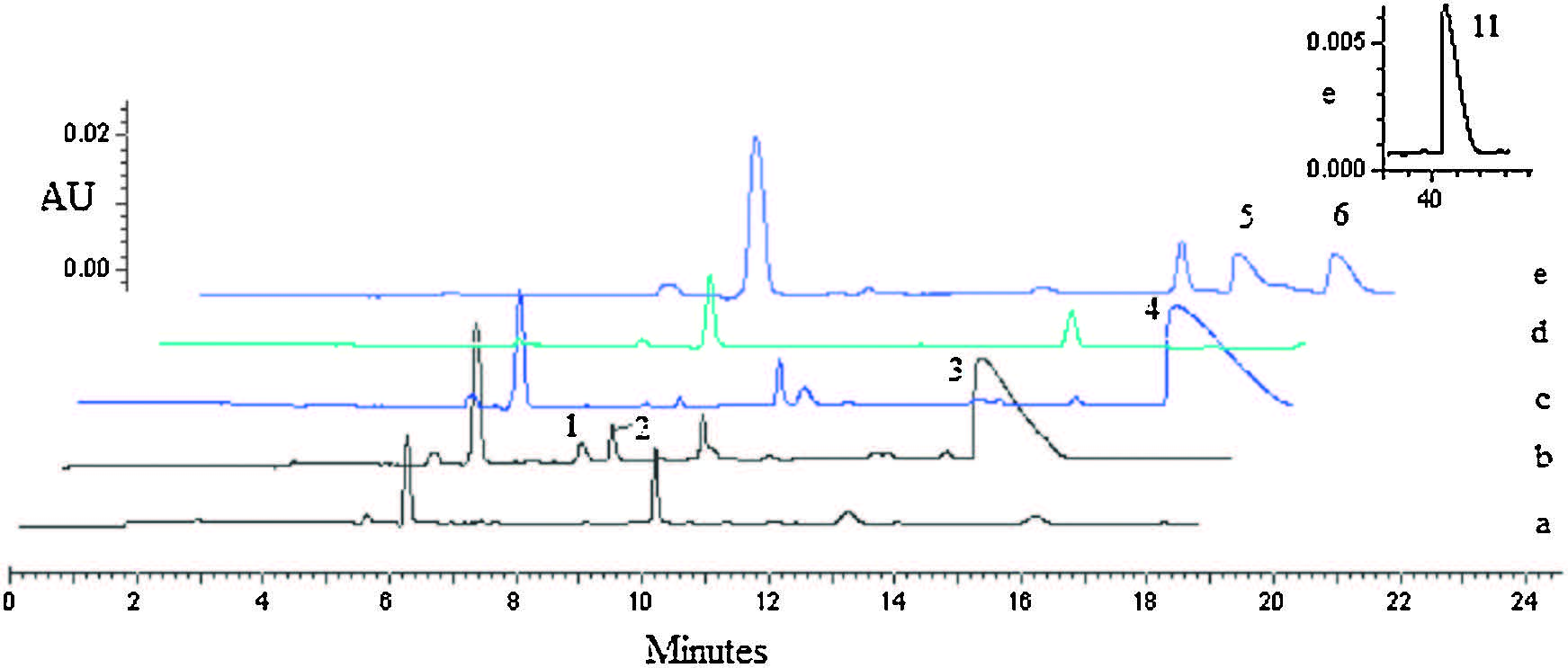
Cytochrome P450 family is an important oxidase system for drug biotransformation and it mainly exists in liver microsome. For all the 6-cyclopropylamino guanosine substrate tested here, after 4 h incubation with mouse liver microsome, D-c-ddG and ABC were converted to the corresponding 6-amino intermediate in varying degrees (Fig. 3, curves b and e). In the incubation solutions of D-cddG and ABC, the respective deamination product of D-ddG and CBV were observed, likely owing to the presence of a certain amount of deaminase in liver microsome.

|
Download:
|
| Figure 3. CE analysis of 6-cyclopropylamino nucleosides incubated in liver microsome individually. a: microsome as blank control to curve b and c; b: D-c-ddG, 4 h; c: L-cddG, 4 h; d: microsome as blank control to curve e; e: ABC, 4 h, ABC was detected at about 42 min and showed on the top of the figure; peak 1: D-a-ddG, peak 2: D-ddG; peak 3: D-c-ddG; peak 4: L-c-ddG; peak 5: a-CBV; peak 6: CBV; peak 7: ABC. | |
{kind=link}
In order to eliminate the effect of deaminase in liver microsome, a deaminase inhibitor, Pentostatin [14], was added into liver microsome incubation solution. No deamination product D-ddG was detected for D-c-ddG when incubated with liver microsome in the presence of Pentostatin (Fig. S3 in Supporting information), which implied that oxidases (CYP) in liver microsome were responsible for the oxidation of 6-cyclopropylamino to produce 6-amino product.
3.3. Deamination in ADA solutionADA was considered as one of the key enzymes for the hydrolytic deamination of purine nucleosides [15]. Previous works had shown that only a tiny portion of D-c-ddG could be metabolized to D-ddG by ADA [16]. Here D/L-forms of 6-cylcopropylamino guanosine analogs were incubated with ADA. We found that D-c-ddG could be partially deaminated to give D-ddG, whereas their L-configuration counterparts could not. ABC was ineffective substrates of ADA (Fig. 4).

|
Download:
|
| Figure 4. CE analysis of 6-cyclpropylamino guanosine analogs treated with ADA. a: D-cddG, 3 h; b: L-c-ddG, 3 h; c: ABC, 3 h; Peak 1: D-ddG; Peak 2: D-c-ddG; Peak 3: L-cddG; Peak 4: ABC. | |
{kind=link}
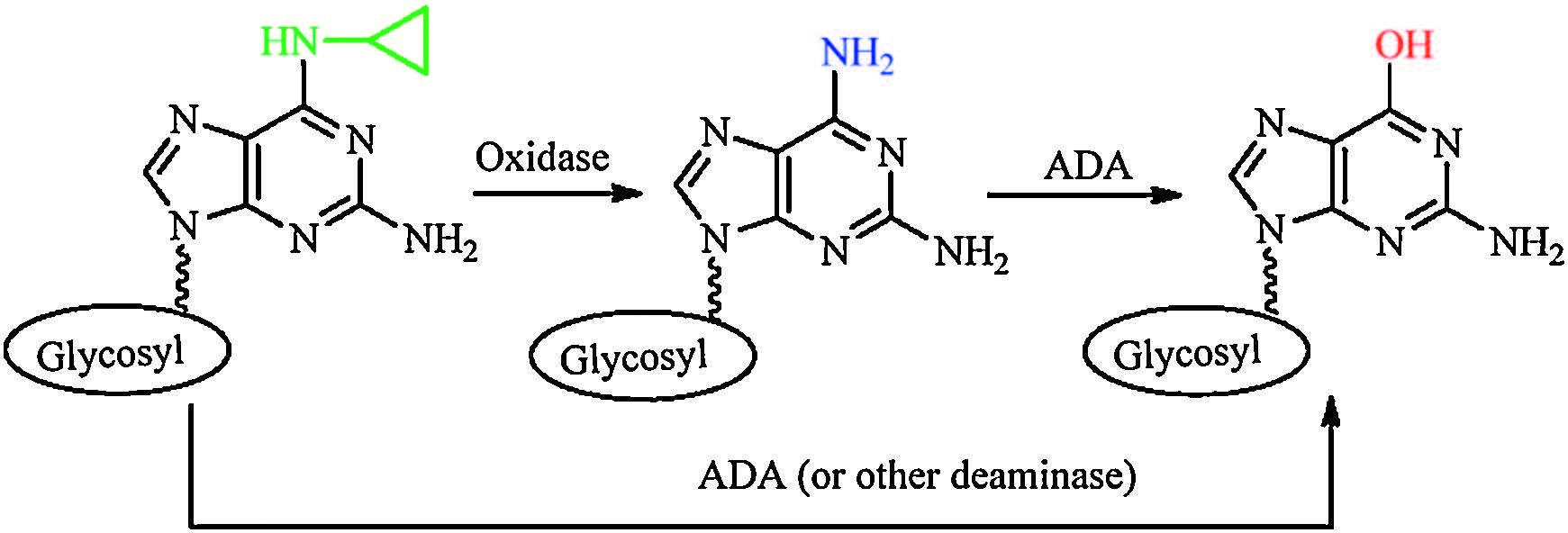
The metabolism study results are summarized in Table 1, which suggested that conversion of 6-cyclopropylamino guanosine analogs in liver might take place through two pathways (Fig. 5). Step 1, enzymatic deamination proceeds stepwise through a 6-amino intermediate, which is formed via oxidation catalyzed by oxidase (CYP), followed by deamination via deaminase (ADA) to produce the guanosine product. During this process, oxidation of the substrates is closely related with the configuration of nucleoside and structure of glycosyl moiety. The subsequent deamination step depends on the configuration of guanosine analogs strictly. Step 2, ADA (or other deaminase) catalyzes deamination to afford guanosine product. For our tested substrates, deamination of L-configuration was much more difficult than that of D-Configuration by mouse liver homogenate/microsome and ADA. It might be due to the specific recognition of nucleoside substrates with different configuration and sugar conformation by the enzymes.

|
Download:
|
| Figure 5. Pathways of conversion of 6-cyclopropylamino guanosine analogs in liver. | |
{kind=link}
4. Conclusion
A CE method was developed for analyzing the metabolic conversion of nucleoside prodrugs in liver. The metabolism of a series of D-/L-6-modified guanosine analogs after treatment with ADA, mouse liver microsome, and liver homogenate were investigated. We confirmed that the metabolism of 6-cyclopropylamino guanosine to guanosine is a two-step process involving 6-amino intermediate formed by oxidative N-dealkylation. The results demonstrated that the conversion of D-/L-6-cyclopropylamino guanosine analogs in liver varied, which was closely related with the configuration of nucleosides and the structure of the glycosyl group. And the remarkable difference of activation for D-/ L-configuration agents highlighted the fact that D-form analogs are much more effective substrates of metabolic enzymes than their Lcounterparts. This finding strongly recommended that modifications of sugar ring or 50-hydroxyl are more feasible than substitution of 6-hydroxyl group in the design of anti-viral agents containing L-guanosine as basic structure.
Acknowledgment The research was supported by National Natural Science Foundation of China (NSFC) (Nos. 21172010,21002004).| [1] | Vince R, Hua M, Brownell J, Potent and selective activity of a new carbocyclic nucleoside analog (carbovir:NSC 614846) against human immunodeficiency virus in vitro. Biochem. Biophys. Res. Commun 156 (1988) 1046–1053. DOI:10.1016/S0006-291X(88)80950-1 |
| [2] | Daluge S.M, Good S.S, Faletto M.B, 1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity. Antimicrob. Agents Chemother 41 (1997) 1082–1093. |
| [3] | Faletto M.B, Miller W.H, Garvey E.P, Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob. Agents Chemother 41 (1997) 1099–1107. |
| [4] | Furman P.A, Jeffrey J, Kiefer L.L, Mechanism of action of 1-β-D-2, 6-diaminopurine dioxolane, a prodrug of the human immunodeficiency virus type 1 inhibitor 1-β-D-dioxolane guanosine. Antimicrob. Agents Chemother 45 (2001) 158–165. DOI:10.1128/AAC.45.1.158-165.2001 |
| [5] | Schinkmanova M, Votruba I, Holy A, N6-methyl-AMP aminohydrolase activates N6-substituted purine acyclic nucleoside phosphonates. Biochem. Pharmacol 71 (2006) 1370–1376. DOI:10.1016/j.bcp.2006.01.013 |
| [6] | Cerny M.A, Hanzlik R.P, Cytochrome P450-catalyzed oxidation of N-benzyl-Ncyclopropylamine generates both cyclopropanone hydrate and 3-hydroxypropionaldehyde via hydrogen abstraction, not single electron transfer. J. Am. Chem. Soc 128 (2006) 3346–3354. DOI:10.1021/ja054938+ |
| [7] | Roberts K.M, Jones J.P, Anilinic N-oxides support cytochrome P450-mediated Ndealkylation through hydrogen-atom transfer. Chemistry 16 (2010) 8096–8107. DOI:10.1002/chem.201000185 |
| [8] | Hostetler K.Y, Richman D.D, Sridhar C.N, Phosphatidylazidothymidine and phosphatidyl-ddC:assessment of uptake in mouse lymphoid tissues and antiviral activities in human immunodeficiency virus-infected cells and in Rauscher leukemia virus-infected mice. Antimicrob. Agents Chemother 38 (1994) 2792–2797. DOI:10.1128/AAC.38.12.2792 |
| [9] | Korba B.A, Xie H, Wright K.N, Liver-targeted antiviral nucleosides:enhanced antiviral activity of phosphatidyl-dideoxyguanosine versus dideoxyguanosine in woodchuck hepatitis virus infection in vivo. Hepatology 23 (1996) 958–963. |
| [10] | Ray A.S, Hernandez-Santiago B.I, Mathew J.S, Mechanism of anti-human immunodeficiency virus activity of beta-D-6-cyclopropylamino-2',3'-didehydro-2',3'-dideoxy-guanosine. Antimicrob. Agents Chemother 49 (2005) 1994–2001. DOI:10.1128/AAC.49.5.1994-2001.2005 |
| [11] | Xie L.J, Yang X.T, Pan D.L, Synthesis and anti-HIV activity of a series of 6-modified 2',3'-dideoxyguanosine and 2',3'-didehydro-2',3'-dideoxyguanosine analogs. Chin. J. Chem 31 (2013) 1207–1218. DOI:10.1002/cjoc.201300440 |
| [12] | Lu J.F, Xie L.J, Cao M, The prodrugs of L-guanosine analogs:design, synthesis and anti-HIV activity. J. Chin. Pharm. Sci 20 (2011) 335–341. |
| [13] | Peng Y, Cheng T, Dong L, Quantification of 2'-deoxy-2'-β-fluoro-4'-azidocytidine in rat and dog plasma using liquid chromatography-quadrupole time-of-flight and liquid chromatography-triple quadrupole mass spectrometry:application to bioavailability and pharmacokinetic studies. J. Pharm. Biomed. Anal 98 (2014) 379–386. DOI:10.1016/j.jpba.2014.06.019 |
| [14] | Frieden C, Kurz L.C, Gilbert H.R, Adenosine deaminase and adenylate deaminase:comparative kinetic studies with transition state and ground state analogue inhibitors. Biochemistry 19 (1980) 5303–5309. DOI:10.1021/bi00564a024 |
| [15] | Wu J.Z, Lin C.C, Hong Z, Ribavirin, viramidine and adenosine-deaminase-catalysed drug activation:implication for nucleoside prodrug design. J. Antimicrob. Chemother 52 (2003) 543–546. DOI:10.1093/jac/dkg405 |
| [16] | Pei L, Xie L.J, Lin Q, Studies on the adenosine deaminase-catalyzed conversion of adenosine and nucleoside prodrugs by different capillary electrophoresis modes. Anal. Biochem 414 (2011) 131–137. DOI:10.1016/j.ab.2011.03.014 |