 2016, Vol. 27
2016, Vol. 27 



b University of Chinese Academy of Sciences, Beijing 100049, China ;
c School of Life Science and Technology, ShanghaiTech University, Shanghai 201210, China
Caesalpinia sappan Linn (Caesalpiniaceae) is a shrubby tree distributed inEast and SoutheastAsia [1-3]. Itsheartwood, containing various structural types of phenolic components such as flavones, homoisoflavonoids, protosappanins and brazilins, has long been used in folk medicines as an antibacterial, anti-inflammatory and analgesic agent [4-7]. Its seeds were reported to be rich source of cassane-type diterpenes [8-14], which are the characteristic natural products of genus Caesalpinia and have shown various kinds of bioactivities, including antimalarial [8, 15], cytotoxic [16, 17], anti-inflammatory [18], antibacterial [9] and antiviral [19-21] activities.
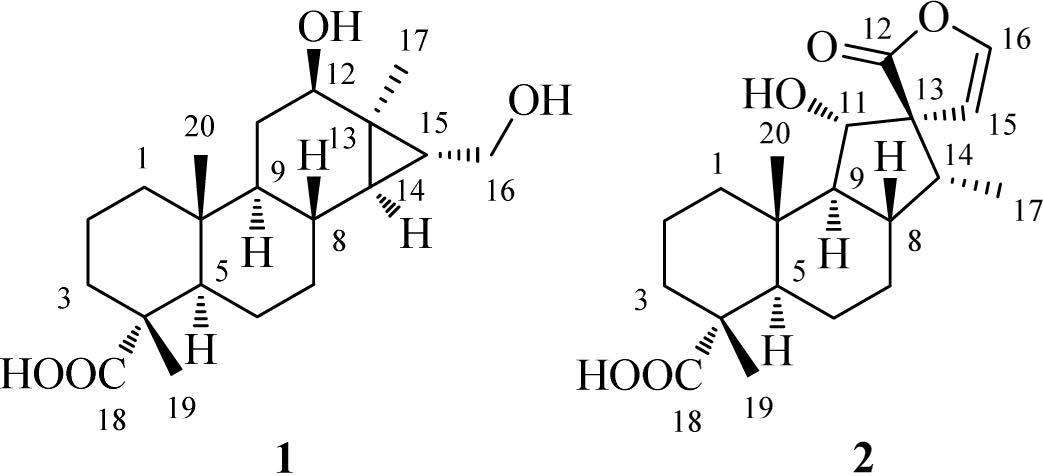
The natural cassane diterpenes are rearranged products of pimarane precursors by amethyl migration fromC-13 to C-14 in the biosynthetic pathway of diterpenes. Although rearranged cassanetype and pimarane-type diterpenes have been reported from the genus Caesalpinia, their number is still a few [13, 16, 22-24]. Our previous study led to the isolation of 10 new cassane diterpenoids from C. bonduc [25]. As a part of our ongoing program toward the discovery of structure-attractive and bioactive compounds fromthe genus Caesalpinia, two new rearranged diterpenoids (Fig. 1) were isolated from the seeds of C. sappan. Among them, tomocinol C (1) showed a rare 6/6/6/3-membered ring system deriving from an ordinary pimarane-type diterpene. Spirocaesalmin C (2), possessing a spiro-heterocyclic skeleton, waspresumably consideredtobe from an ordinary cassane-type diterpene. Herein, we report the isolation and structure elucidation of compounds 1 and 2.

|
Download:
|
| Figure 1. Structures of compounds 1 and 2. | |
2. Experimental 2.1. General experimental procedures
Optical rotation data were obtained using a Rudulph Autopol VI Automatic polarimeter. UV data were recorded with a Shimadzu UV-2550 spectrometer. IR data were measured on a Nicolet Magna FTIR-750 spectrometer using KBr disks. NMR spectra were recorded on a Bruker AM-400 (Bruker, Ettlingen, Germany) and a Bruker Avance III (Bruker, Ettlingen, Germany) 500 NMR spectrometer with TMS as internal standard. HRESIMS spectra were detected on an Agilent G6224A TOF spectrometer. Analytical HPLC and ESIMS spectra were performed on a Waters 2695 instrument with a 2998 PDA coupled with a Waters Acquity ELSD and a Waters 3100 SQDMS detector. TLC was performed on pre-coated silica gel GF254 plates (QingDao Marine Chemical Industrials). MCI gel CHP20P (75-150 mm, Mitsubishi Chemical Industries, Japan), Silica gel (Qingdao Marine Chemical Industrials), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used for column chromatography (CC). Preparative HPLC was carried out on a Varian PrepStar system with an Alltech 3300 ELSD using a Waters Sunfire RP C18 column (5 μm, 30 mm - 150 mm). All solvents used for CC and HPLC were of analytical grade (Shanghai Chemical Reagents Co., Ltd.) and gradient grade (Merck KGaA, Germany), respectively.
2.2. Plant materialThe seeds of C. sappan were collected in Zhanjiang City, Guangdong Province, China, in December 2014, and were identified by Prof. Jin-Gui Shen from the Shanghai Institute of Materia Medica. A voucher specimen (No. 20141213) has been deposited at the Herbarium of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
2.3. Extraction and isolationThe air-dried, powdered seeds of C. sappan (11 kg) were extracted with 95% EtOH (3×30 L) at room temperature (72 h each), and the combined EtOH extracts were then concentrated under reduced pressure to give a crude residue (1.05 kg). The residue was suspended in water and then partitioned with petroleum ether (PE) and EtOAc successively, affording a PE extract (135 g) and a EtOAc extract (140 g), respectively. The EtOAc extract was chromatographed on a MCI column eluted with EtOH in water in a step gradient manner (25, 50, 80 and 95%) to afford four fractions (Fr.1-Fr.4). Fr.2 was fractionated on silica gel CC (200-300 mesh) eluted gradiently with PE-acetone (8:1 to 0:1) to afford seven subfractions (Fr.2A-Fr.2G). Fr.2F was subjected to a silica gel column (200-300 mesh) eluted gradiently with CH2Cl2-MeOH (40:1 to 10:1) to provide four subfractions (Fr.2F1-Fr.2F4). Fr.2F2 was further purified by Sephadex LH-20 gel CC eluted with MeOH to afford 1 (67 mg). Fr.3 was subjected to a silica gel column (200-300 mesh) eluted gradiently with PE-acetone (8:1 to 0:1) to provide ten subfractions (Fr.3A-Fr.3 J). J). Fr.3E was separated by silica gel CC (200-300 mesh) eluted gradiently with CH2Cl2-MeOH (200:1 to 40:1) to obtain three subfractions (Fr.3E1-Fr.3E3). Fr.3E3 was further purified with preparative HPLC (MeCN-H2O, 30%-45%, 0-100 min, 25.0 mL/ min) to yield 2 (39 mg).
Tomocinol C (1): Colorless crystal; [a]D24 +26.0 (c 0.2, MeOH); IR (KBr, cm-1): vmax 3420, 2925, 1726, 1701; 1H NMR and 13C NMR data, see Table 1; ESIMS m/z 335.2 [M-H]-; HRESIMS m/z 359.2202 [M + Na]+ (calcd. for C20H32O4Na, 359.2194).
|
|
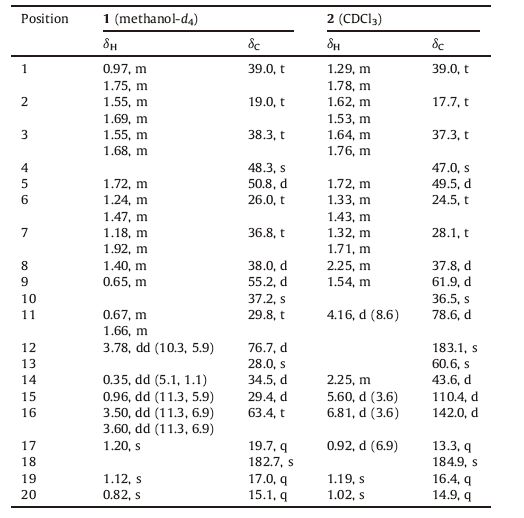
Table 1 1H NMR (400 MHz, d in ppm, J in Hz) and 13C NMR (125 MHz, δ in ppm) data for compounds 1 and 2. |
{kind=link}
Spirocaesalmin C (2): Colorless crystal; [a]D24 +39.3 (c 0.2, CHCl3); IR (KBr, cm-1): vmax 3443, 3267, 2948, 1758, 1700; 1HNMR and 13C NMR data, see Table 1; ESIMS m/z 347.2 [M-H]-; HRESIMS m/z 371.1833 [M + Na]+ (calcd. for C20H28O5Na, 371.1830).
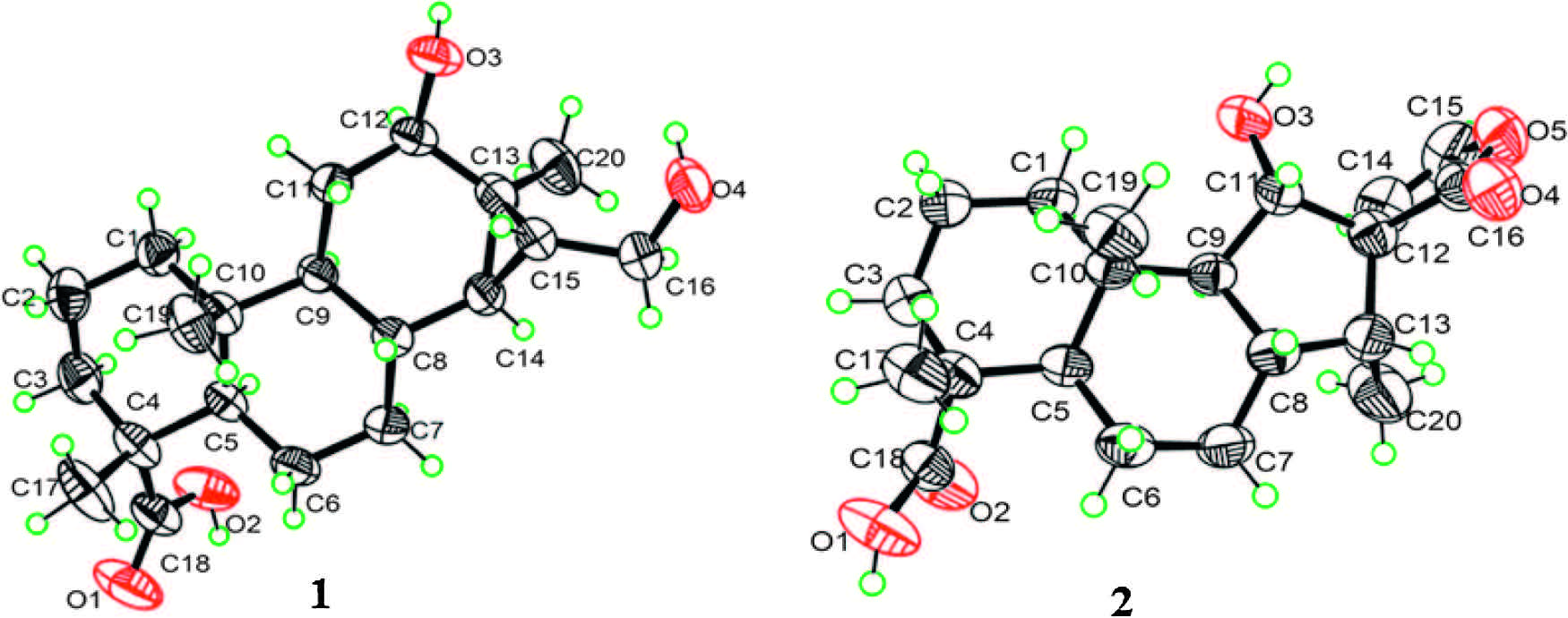
2.4. Crystal data of 1 and 2Crystallographic data for 1 (CCDC 1450762) and 2 (CCDC 1450763) are attached in the Supporting information (Fig. S9 and Fig. S18 in Supporting information). Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB21EZ, UK.
3. Results and discussionCompound 1, isolated as colorless crystals frommethanol, had a molecular formula of C20H32O4 as determined by the pseudomolecular ion peak at m/z 359.2202 [M+ Na]+ in HRESIMS with five degrees of unsaturation. The IR absorption bands at 3420 and 1726 cm-1 indicated the presence of hydroxyl and carbonyl groups, respectively. The 1H NMR (Table 1) spectrum displayed signals attributable to three tertiary methyls (δH 0.82, 1.12, 1.20), an oxygenated methine (δH 3.78, dd, J = 10.3, 5.9), and an oxygenated methylene (δH 3.50, dd, J = 11.3, 6.9; δH 3.60, dd, J = 11.3, 6.9). All carbon signalswerewell resonated in the 13CNMRandDEPT spectra (Table 1), and were classified as three methyls, seven methylenes (one oxygenated), six methines (one oxygenated) and four quarternary carbons (one carboxyl). To accommodate seven degrees of unsaturation implied by themolecular formula, compound 1 was proposed to be a tetracyclic diterpenoid. The proton signals were assigned to the corresponding carbons through direct 1H and 13C correlations in the HSQC spectrum. The 1H-1H COSY data (Fig. 2) enabled the construction of the partial structures depicted by the bold lines, whichwere connected through analysis of theHMBC data (Fig. 2). The obvious upfield shift of two methines (δH 0.35, δC 34.5, CH-14; δH 0.96, δC 29.4, CH-15) and a quaternary carbon (δC 28.0, C-13), together with the HMBC correlations from the oxygenated methylene (H2-16) to C-13, C-14, and C-15, suggested that C-13, C-14 and C-15 formed a cyclopropane ring with a hydroxymethyl attached to C-15. The hydroxyl group was assigned at C-12 on the basis of the HMBC correlations from H-9 and H2-11 to C-12. The observed HMBC correlations from a tertiary methyl at δH 1.20 (H3-17) to C-12 and C-15 confirmed its location at C-13. In addition, theHMBCcorrelations fromH3-19toC-3, C-5anδC-18, andfromH3- 20 to C-1, C-5 and C-9 were observed, which proved the locations of twomethyls. The planar structure of 1was eventually established as a 6/6/6/3-membered ring system(Fig. 1). The relative structure of 1 was determined by theROESY experiment (Fig. 2). The cross peaks of H3-19/H3-20, H3-20/H-8, H-8/H-15, H-5/H-9, H-9/H-12, H-14/H2-16 and H2-16/H3-17 suggested that CH3-19, CH3-20, H-8 and OH-12 were b-oriented, while C-18, H-5, H-9, H-12, H-14, H2-16 and H3-17 werea-oriented.Asingle crystal X-ray diffraction experimentwith a Cu radiation source confirmed further the whole structure of 1 including the absolute configuration (Fig. 3).Therefore, the structure of 1 was established as shown and named tomocinol C.

|
Download:
|
| Figure 2. The 1H-1H COSY (bold), HMBC (!), and ROESY correlations of compounds 1 and 2. | |
{kind=link}

|
Download:
|
| Figure 3. X-ray crystallographic structures of compounds 1 and 2. | |
{kind=link}
Compound 2, colorless crystals from acetone, exhibited a pseudo-molecular ion peak [M + Na]+ at m/z 371.1833 in the HRESIMS, corresponding to a molecular formula of C20H28O5. The IR spectrum indicated the occurrence of hydroxyl (3443 cm-1) and carbonyl (1758 cm-1) groups. The 1H NMR (Table 1) spectrum indicated the presence of two tertiary methyls (δH 1.02, 1.19), a secondary methyl (δH 0.92, d, J = 6.9), an oxygenated aliphatic methine (δH 4.16, d, J = 8.6) and two protons (δH 5.60, d, J = 3.6; δH 6.81, d, J = 3.6) of a 1, 2-disubstituted furan ring. The 13C NMR spectrum (Table 1) exhibited 20 resonances including three methyls, five methylenes, seven methines (one oxygenated and two sp2) and five quaternary carbons (two carboxyls). The carbonbonded protons of 2 were assigned from the HSQC spectrum. Analysis of the 1H-1H COSY spectrum (Fig. 2) led to the partial structures depicted by the bold lines, which were connected on the basis of the long-range correlations observed in the HMBC spectrum (Fig. 2). The hydroxyl group was designated at C-11 based on the HMBC correlation from H-11 (δH 4.16, d, J = 8.6) to C-9 (δC 61.9) and C-10 (δC 36.5). The presence of a spiro-heterocyclic moiety was determined by the HMBC correlations from H-11, H- 14, H-15, H-16 and H3-17 to the quaternary C-13, and from H-14, H-15 and H-16 to C-12. Furthermore, the HMBC correlations from H3-19 to C-3, C-4, C-5 and C-18, and from H3-20 to C-1, C-5, C-9 and C-10 confirmed the connection of the rest part of the structure. Thus, the planar structure of 2 was thus constructed. The relative configuration of 2 was determined by the ROESY correlations of H3-19/H3-20, H3-20/H-8, H3-20/H-11, H-11/H-14 and H-15/H3-17 (Fig. 2). The proposed structure of 2 including the absolute configuration was finally confirmed by a single crystal X-ray experiment (Fig. 3). Accordingly, the structure of 2 was established as shown and named spirocaesalmin C.
Compounds 1 and 2 were tested their inhibitory effects against Plasmodium falciparum 3D7 asexual stage malaria parasites, which is a drug sensitive strain. Two independent in vitro assays were carried out, and the data are given as inhibition (mean% - SD) at 10 mmol/L. The results showed that both 1 and 2 were inactive against 3D7 strain of P. falciparum at the concentration of 10 mmol/L.
4. ConclusionTwo rearranged diterpenes, tomocinol C(1) andspirocaesalmin C (2), were fully characterized fromthe seeds of C. sappan. TomocinolC (1) exhibited a rare carbon framework with a 6/6/6/3-membered ring system, which was proposed to be rearranged froma pimarane diterpenoid by forming a new linkage between C-14 and C-15. Spirocaesalmin C (2), featuring by a spiro-heterocyclic structure, was probably a rearranged product of a cassane diterpene with a migration of the C-11 fromC-12 to C-13. Both skeletons were rarely found, with only a few literatures reported.
Acknowledgments Weare thankful for the financial support of the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (No. 2012ZX09301001-001). Our thanks are also given to the National Natural Science Funds (No. 81302657,81473112,81573305) and for grants from the Chinese Academy of Sciences (No. KSZD-EW-Z-004-01),and the Shanghai Commission of Science and Technology (No. 12JC1410300).| [1] | Wang Z, Sun J.B, Qu W, Caesappin A and B, two novel protosappanins from Caesalpinia sappan L. Fitoterapia 92 (2014) 280–284. DOI:10.1016/j.fitote.2013.12.004 |
| [2] | Nirmal N.P, Rajput M.S, Prasad R.G, Brazilin from Caesalpinia sappan heartwood and its pharmacological activities:a review. Asian Pac. J. Trop. Med 8 (2015) 421–430. DOI:10.1016/j.apjtm.2015.05.014 |
| [3] | Zhang J.Y, Wu F.H, Qu W, Two new cassane diterpenoids from the seeds of Caesalpinia sappan Linn. Chin. J. Nat. Med 10 (2012) 218–221. DOI:10.3724/SP.J.1009.2012.00218 |
| [4] | Ou Yang B, Ke C.Q, He Z.S, Brazilide A, a novel lactone with an unprecedented skeleton from Caesalpinia sappan. Tetrahedron Lett 43 (2002) 1731–1733. DOI:10.1016/S0040-4039(02)00109-0 |
| [5] | Nguyen M.T.T, Awale S, Tezuka Y, Xanthine oxidase inhibitors from the heartwood of Vietnamese Caesalpinia sappan. Chem. Pharm. Bull 53 (2005) 984–988. DOI:10.1248/cpb.53.984 |
| [6] | Cuong T.D, Hung T.M, Kim J.C, Phenolic compounds from Caesalpinia sappan heartwood and their anti-inflammatory activity. J. Nat. Prod 75 (2012) 2069–2075. DOI:10.1021/np3003673 |
| [7] | Shu S.H, Deng A.J, Li Z.H, Two novel biphenyl dimers from the heartwood of Caesalpinia sappan. Fitoterapia 82 (2011) 762–766. DOI:10.1016/j.fitote.2011.03.010 |
| [8] | Ma G, Wu H, Chen D, Antimalarial and antiproliferative cassane diterpenes of Caesalpinia sappan. J. Nat. Prod 78 (2015) 2364–2371. DOI:10.1021/acs.jnatprod.5b00317 |
| [9] | Zhang J, Abdel-Mageed W.M, Liu M, Caesanines A-D, new cassane diterpenes with unprecedented N bridge from Caesalpinia sappan. Org. Lett 15 (2013) 4726–4729. DOI:10.1021/ol402058z |
| [10] | Nguyen H.X, Nguyen N.T, Dang P.H, Cassane diterpenes from the seed kernels of Caesalpinia sappan. Phytochemistry 122 (2016) 286–293. DOI:10.1016/j.phytochem.2015.12.018 |
| [11] | Tran M.H, Nguyen M.T, Nguyen H.D, Cytotoxic constituents from the seeds of Vietnamese Caesalpinia sappan. Pharm. Biol 53 (2015) 1549–1554. DOI:10.3109/13880209.2014.986686 |
| [12] | Wu H.F, Zhu Y.D, Sun Z.H, Norcassane- and cassane-type furanoditerpenoids from the seeds of Caesalpinia sappan. Fitoterapia 98 (2014) 22–26. DOI:10.1016/j.fitote.2014.07.001 |
| [13] | Yodsaoue O, Cheenpracha S, Karalai C, Phanginin A-K, diterpenoids from the seeds of Caesalpinia sappan Linn. Phytochemistry 69 (2008) 1242–1249. DOI:10.1016/j.phytochem.2007.11.013 |
| [14] | Ma G.X, Zhu Y.D, Sun Z.H, Three new cassane diterpenes from the seeds of Caesalpinia sappan. Phytochem. Lett 8 (2014) 141–144. DOI:10.1016/j.phytol.2014.03.008 |
| [15] | Kalauni S.K, Awale S, Tezuka Y, Antimalarial activity of cassane- and norcassane-type diterpenes from Caesalpinia Crista and their structure-activity relationship. Biol. Pharm. Bull 29 (2006) 1050–1052. DOI:10.1248/bpb.29.1050 |
| [16] | Zheng Y, Zhang S.W, Cong H.J, Caesalminaxins A-L, cassane diterpenoids from the seeds of Caesalpinia minax. J. Nat. Prod 76 (2013) 2210–2218. DOI:10.1021/np400545v |
| [17] | Ma G, Yuan J, Wu H, Caesalpins A-H, bioactive cassane-type diterpenes from the seeds of Caesalpinia minax. J. Nat. Prod 76 (2013) 1025–1031. DOI:10.1021/np300918q |
| [18] | Dong R, Yuan J, Wu S, Anti-inflammation furanoditerpenoids from Caesalpinia minax Hance. Phytochemistry 117 (2015) 325–331. DOI:10.1016/j.phytochem.2015.06.025 |
| [19] | Jiang R.W, Ma S.C, He Z.D, Molecular structures and antiviral activities of naturally occurring and modified cassane furanoditerpenoids and friedelane triterpenoids from Caesalpinia minax. Bioorg. Med. Chem 10 (2002) 2161–2170. DOI:10.1016/S0968-0896(02)00072-X |
| [20] | Jiang R.W, But P.P.H, Ma S.C, Structure and antiviral properties of macrocaesalmin, a novel cassane furanoditerpenoid lactone from the seeds of Caesalpinia minax Hance. Tetrahedron Lett 43 (2002) 2415–2418. DOI:10.1016/S0040-4039(02)00232-0 |
| [21] | Jiang R.W, Ma S.C, But P.P.H, New antiviral cassane furanoditerpenes from Caesalpinia minax. J. Nat. Prod 64 (2001) 1266–1272. DOI:10.1021/np010174+ |
| [22] | Yodsaoue O, Karalai C, Ponglimanont C, Pulcherrins D-R, potential antiinflammatory diterpenoids from the roots of Caesalpinia pulcherrima. Tetrahedron 67 (2011) 6838–6846. DOI:10.1016/j.tet.2011.06.087 |
| [23] | Zhang J.L, Tian H.Y, Chen N.H, Caesalpinimin A, a novel rearranged furanoditerpene with an unprecedented carbon skeleton from the seeds of Caesalpinia minax Hance. RSC Adv 4 (2014) 7440–7443. DOI:10.1039/c3ra46502k |
| [24] | Wu J, Chen G, Xu X, Seven new cassane furanoditerpenes from the seeds of Caesalpinia minax. Fitoterapia 92 (2014) 168–176. DOI:10.1016/j.fitote.2013.11.002 |
| [25] | Zhang P, Tang C, Yao S, Cassane diterpenoids from the pericarps of Caesalpinia bonduc. J. Nat. Prod 79 (2016) 24–29. DOI:10.1021/acs.jnatprod.5b00520 |