 2016, Vol. 27
2016, Vol. 27 



Isatis in digotica Fort. (Cruciferae) is a cultivated medicinal plant. Its dried leaves and roots, named “da qing ye” and “ban lan gen” in Chinese, respectively, are common ingredients of formulations for the treatment of influenza and other infections [1, 2]. Previous pharmacological and chemical studies indicated that extracts of “da qing ye” and “ban lan gen” displayed similar activities and contained similar constituents [3-5]. There were reports that the “da qing ye” extract was more potent than the “ban lan gen” extract [6] and that the former contained structurally unique constituents [7, 8]. Especially, a literature survey indicated that about 40 chemical constituents were reported from “da qing ye” and that the previous studies were mainly carried out on the ethanol and methanol extracts of the drug materials, which differs from practical application of water decoctions. Therefore, as part of a program to systematically assess the chemical and biological diversity of traditional Chinese medicines, focusing on the minor components [9-23], we conducted investigations of the aqueous extracts of “ban lan gen” and “da qing ye”, respectively. From the “ban lan gen” extract, 45 new alkaloids, including a pair of indole alkaloid enantiomers containing dihydrothiopyran and 1, 2, 4-thiadiazole rings, a pair of bisindole alkaloid enantiomers, four stereoisomers of 3, 5-bis(2-hydroxybut-3-en-π*1-yl)-1, 2, 4-thiadiazole, and 12 glycosidic indole and bisindole alkaloids [24-32]. Some of these compounds showed antiviral activity against influenza virus A/Hanfang/359/95 (H3N2) or Coxsackie virus B3 and protective activity against dl-galactosamine-induced hepatocyte (WB-F344 cell) damage. From the “da qing ye” aqueous extract, herein, we report details of the isolation and structure elucidation of two pairs of unusual scalemic stereoisomers of 2-[10- (4"-hydroxy-3", 5"-dimethoxyphenyl)ethyl]-2-methoxyindolin-3- one, named isatidifoliumindolinones A-D (1-4, Fig. 1).

|
Download:
|
| Figure 1. Structures of 1-4. | |
2. Experimental 2.1. General experimental procedures
Optical rotations were measured on a P-2"0 polarimeter. UV spectra were recorded on a V-650 spectrometer. CD spectra were measured on a JASCO J-815 CD spectrometer. IR spectra were recorded on a Nicolet 57" FT-IR Microscope spectrometer (FT-IR Microscope Transmission). 1D- and 2D-NMR spectra were obtained at 6" for 1H and 150 MHz for 13C, respectively, on an Inova 6" MHz spectrometer with solvent peaks as references. ESIMS and HR-ESIMS data were obtained on an AccuToFCS JMST1"CS spectrometer. Column chromatography (CC) was performed with HPD-110 (Cangzhou Bon Absorber Technology Co.Ltd., Cangzhou, China), MCI gel CHP 20P (Mitsubishi Chemical Inc., Tokyo, Japan), silica gel (2"-3" mesh, Qingdao Marine Chemical Inc., China), RP silica gel (Grace Davison Discovery Science, Deerfield, USA), or Toyopearl HW-40C gel (Tosoh Corporation Bioscience Davison, Tokyo, Japan). HPLC separation was performed on a CXTH3"0 system (Beijing Tong Heng Innovation Technology Co., Ltd., Beijing, China) with a semi-preparative column COSMOSIL (250 mm - 10 mm i.d.) packed with C18 silica gel (5 mm) and a semi-preparative chiral column (Chiralpak AD-H, 250 mm - 10 mm) packed with amylose tris-(3, 5-dimethylphenylphenylcarbamate) coated on 5 mm silica-gel (Daicel Chiral Technologies Co., Ltd., Shanghai, China). TLC was conducted on precoated silica gel GF254 plates (Yantai Chemical Industry Research Institute, Yantai, China). Spots were visualized under UV light (254 or 365 nm) or by spraying with 10% H2SO4 in 95% EtOH followed by heating.
2.2. Plant materialDa qing ye (the dried leaves of I. in digotica) were purchased in Beijing Tong Ren Tang drugstore, which was collected in Hebei Province, China, in December 2013. The plant material was identified by Mr. Lin Ma (Institute of Materia Medica, Beijing 1"050, China). A voucher specimen (No. ID-S-2546) was deposited at the Herbarium of the Department of Medicinal Plants, Institute of Materia Medica, Beijing, China.
2.3. Extraction and isolationThe pulverized plant material (1" kg) was decocted with H2O (1"0 L; 3- 0.5 h). The aqueous extracts were combined and evaporated under reduced pressure to yield a dark-brown residue (42 kg). The residue was dissolved in H2O (50 L), loaded on a macroporous adsorbent resin (HPD-110, 19 kg) column (20 cm - 2" cm), and eluted successively with H2O (2" L), 50% EtOH (425 L), and 95% EtOH (250 L) to yield three corresponding fractions A, B and C. After removing the solvent under reduced pressure, fraction B (3.7 kg) was separated by column chromatography (CC) over MCI gel CHP 20P (20 L), with successive elution using H2O (2" L), 30% EtOH (1" L), 50% EtOH (1" L), 95% EtOH (80 L), and Me2CO (30 L), to give fractions B1-B5. Fraction B2 (1.72 kg) was subjected to CC over silica gel (5 kg), with elution by a gradient of increasing MeOH concentration (0-1"%) in EtOAc and then with H2O, to yield subfractions B2-1-B2-10 based on TLC analysis. Subfraction B2-4 (2" g) was chromatographed over silica gel (2 kg), eluting with CH2Cl2-CH3OH (13:1) then MeOH, to yield fractions B2-4-1-B2-4-6, of which B2-4-2 (20 g) was further fractionated by CC over RP (C18) silica gel (36 mm - 460 mm, 150 g) eluting with a gradient of increasing MeOH concentration (0-1"%) in H2O to yield B2-4-2-1-B2-4-2-28. Fraction B2-4-2-26 (60 mg) was separated by CC over HW-40C, eluting with MeOH, followed by RP HPLC (C18 column, 52% MeOH in H2O, flow rate 2.0 mL/min), to yield mixtures 1 and 2 (6.5 mg, tR = 38.6 min) and 3 and 4 (6.0 mg, tR = 23.5 min). Compounds 1 (2.0 mg, tR = 26.1 min) and 2 (1.2 mg, tR = 32.6 min) were separated by chiral HPLC with a Chiralpak AD-H column (250 mm - 10 mm) using a mobile phase of EtOH-n-π*hexane (1:4, flow rate 2.5 mL/min), while 3 (2.0 mg, tR = 21.6 min) and 4 (1.2 mg, tR = 18.6 min) were separated with the same chiral column using the mobile phase of EtOH-n-π*hexane (3:7, flow rate 1.8 mL/min).
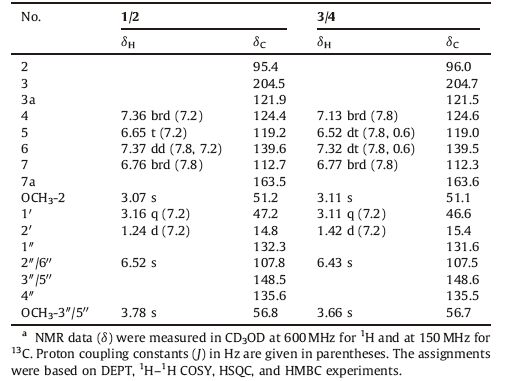
Isatidifoliumindolinone A (1): Yellowish amorphous powder; [a]D 20 -32.6 (c 0.05, MeOH); UV (MeOH) λmax (log e): 206 (3.93), 234 (3.71), 265 (3.10, sh), 410 (2.67) nm; CD (MeOH) 224 (Δε- 2.81), 259 (sh, Δε- 0.83), 295 (sh, Δε- 0.28), 347 (Δε+ 2.57), 418 (Δε- 1.") nm; IR vmax 3531, 3360, 2928, 2846, 1701, 1620, 1519, 1491, 1463, 1429, 1376, 1324, 1301, 1209, 1114, 1052, 987, 908, 844, 803, 779, 749, 706, 695, 656, 595, 526 cm-1; 1H NMR (CD3OD, 6" MHz) data, see Table 1; 13C NMR (CD3OD, 150 MHz) data, see Table 1; (+)-ESIMS m/z 366 [M+Na]+; (+)-HR-ESIMS m/z 366.1308 [M+Na]+ (calcd. for C19H21NO5Na, 366.1312).
|
|
Table 1 NMR spectroscopic data for 1-4 in CD3ODa. |
{kind=link}
Isatidifoliumindolinone B (2): Yellowish amorphous powder; [a]D 20 +28.0 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 206 (3.96), 234 (3.74), 265 (3.10, sh), 411 (2.74) nm; CD (MeOH) 223 (Δε+ 2.89), 259 (sh, Δε+ 0.70), 295 (sh, Δε+ 0.27), 347 (Δε- 2.48), 420 (Δε+ 1.01) nm; IR vmax 3529, 3363, 2930, 2846, 1701, 1622, 1519, 1490, 1467, 1428, 1323, 1214, 1117, 1064, 986, 908, 844, 803, 778, 750, 706, 694, 655, 595, 526 cm-1;1H NMR (CD3OD, 6" MHz) data, see Table 1; 13C NMR (CD3OD, 150 MHz) data, see Table 1; (-)-ESIMS m/z 342 [M-H]-; (+)-HR-ESIMS m/z 366.1312 [M+Na]+ (calcd. for C19H21NO5Na, 366.1312).
Isatidifoliumindolinone C (3): Yellowish amorphous powder; [a]D 20 -70.1 (c 0.10, MeOH); UV(MeOH) λmax (log ε) 206 (3.93), 233 (3.68), 266 (2.99, sh), 410 (2.70) nm; CD (MeOH) 221 (Δε+ 5.25), 264 (Δε+ 1.88), 294 (sh, Δε+ 0.70), 346 (Δε- 2.78), 430 (Δε+ 0.40) nm; IR vmax 3363, 2936, 2840, 1699, 1619, 1519, 1489, 1464, 1429, 1370, 1324, 1217, 1152, 1116, 1025, 988, 903, 839, 801, 755, 712, 656, 587, 531 cm-1; 1H NMR (CD3OD, 6" MHz) data, see Table 1; 13C NMR (CD3OD, 150 MHz) data, see Table 1; (+)-ESIMS m/z 366 [M+Na]+; (+)-HR-ESIMS m/z 366.1310 [M+Na]+ (calcd for C19H21NO5Na, 366.1312).
Isatidifoliumindolinone D (4): Yellowish amorphous powder; [a]D 20 +68.0 (c 0.10, MeOH); UV(MeOH) λmax (log e) 206 (3.93), 233 (3.67), 264 (3.04, sh), 409 (2.71) nm; CD (MeOH) 222 (Δε- 6.50), 264 (Δε- 1.95), 294 (sh, Δε- 0.71), 346 (Δε+ 3.16), 429 (Δε- 0.36) nm; IR vmax 3367, 2970, 2938, 2840, 1697, 1619, 1519, 1490, 1464, 1429, 1370, 1324, 1218, 1117, 1045, 988, 903, 839, 801, 755, 712, 656, 587, 536 cm-1; 1H NMR (CD3OD, 6" MHz) data, see Table 1; 13C NMR (CD3OD, 150 MHz) data, see Table 1; (+)-ESIMS m/z 366 [M + Na]+; (+)-HR-ESIMS m/z 366.1315 [M+Na]+ (calcd. for C19H21NO5Na, 366.1312).
2.4. ECD calculation of stereoisomers (1-4)Conformational analysis of (1′R, 2R)- or (1′R, 2S)-isomer (1 or 3) was conducted by Monte Carlo searching with the MMFF94 molecular mechanics force field using the MOE software package [33]. The lowest-energy conformers having relative energies within 2 kcal/mol were optimized with the Gaussian 09 program [34]. Subsequently, the conformers were optimized using DFT at the B3LYP/6-31+G (d, p) level (Tables S1 and S2 and Figs. S1 and S2 in Supporting information), with the solvent effects considered using the dielectric constant of MeOH (e = 32.6) via conductor-like polarizable continuum model (CPCM). The energies, oscillator strengths, and rotational strengths of the first 60 electronic excitations were calculated using the TDDFT methodology at the B3LYP/6-311++G (2d, 2p) level in vacuum. The re-optimized conformers showed relative Gibbs free energies (ΔG) under 2 kcal/mol were used for ECD spectra simulation. The ECD spectra were simulated by the Gaussian function (s = 0.28 eV). To obtain the final spectra of (1′R, 2R)- or (1′R, 2S)-isomer, the simulated spectra of the lowest energy conformations were averaged on the basis of the Boltzmann distribution theory and their relative Gibbs free energy (ΔG). The corresponding theoretical ECD spectrum of (1′S, 2S)- or (1′S, 2R)-isomer (2 or 4) was obtained by inverting that of (1′R, 2R)- or (1′R, 2S)-isomer (Fig. S3 in Supporting information).
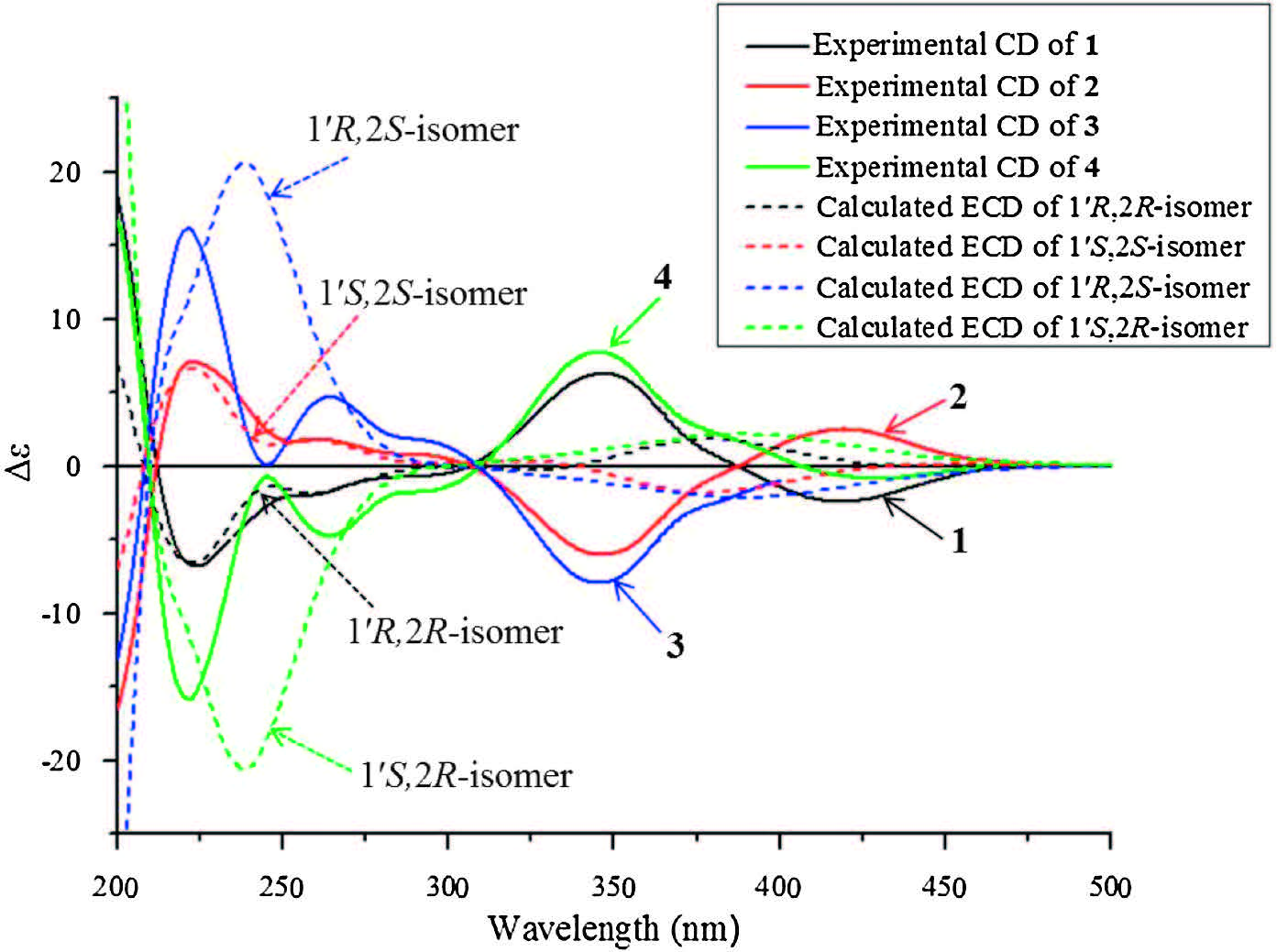
3. Results and discussionCompound 1 was obtained as a yellowish amorphous powder with [a]D 20 -32.6 (c 0.05, MeOH). Its IR spectrum showed absorptions for hydroxy (3360 cm-1), conjugated carbonyl (1701 cm-1), and aromatic ring (1620, 1519, and 1491 cm-1) functional groups. The positive mode ESIMS of 1 exhibited a quasimolecular ion peak at m/z 366 [M+Na]+, and the molecular formula was determined as C19H21NO5 by HR-ESIMS at m/z 366.1308 [M+Na]+ (calcd. for C19H21NO5Na, 366.1312), combined with the NMR data (Table 1). The 1H NMR spectrum of 1 exhibited resonances attributable to an ortho-disubstituted benzene ring at δH 7.36 (brd, J = 7.2 Hz, H-4), 6.65 (t, J = 7.2 Hz, H-5), 7.37 (dd, J = 7.8, 7.2 Hz, H-6) and 6.76 (brd, J = 7.8 Hz, H-7); a 1′, 1′-disubstituted ethyl unit at δH 3.16 (q, J = 7.2 Hz, H-1′) and 1.24 (d, J = 7.2Hz, H3- 20); a 4"-hydroxy-3", 5"-dimethoxyphenyl at δH 3.78 (6H, s, OCH3- 3", 5") and 6.52 (2H, s, H-2"/6") [35]; and a methoxy group at δH 3.07 (s, OCH3-2). The 13C NMR and DEPT spectra displayed signals (Table 1) corresponding to the above units and two additional quaternary carbons attributable to ketal (δC 95.4, C-2) and ketone (δC 204.5, C-3) carbons, respectively. As compared with those of the previously isolated constituents from the extracts of this plant [24-32], these spectroscopic data suggest that 1 possesses an unusual structural feature, which was further elucidated by 2D NMR data. The proton resonances and corresponding protonbearing carbon resonances in theNMR spectrawere unambiguously assigned by HSQC spectroscopic data analysis. Homonuclear vicinal coupling correlations of H-4/H-5/H-6/H-7 in the 1H-1H COSY and heteronuclear three-bond correlations from H-4 to C-3, C-6, and H- 7a; fromH-5 to C-3a and C-7; fromH-6 to C-4 and C-7a; and fromH- 7 to C-5 and C-3a in theHMBC (Fig. 2); together with chemical shifts of these proton and carbon resonances, revealed the presence of an ortho-substituted benzoyl moiety in 1. The 1H-1H COSY cross peaks between H-1′ and H3-20 and HMBC correlations from H-10 to C-1" and C-2"/6"; fromH3-20 to C-1"; fromH-2"/6" to C-10, C-3"/5", and C-4"; from OCH3-3"/5" to C-3"/5"; combined with their chemical shifts, indicated that there was a 10-(4"-hydroxy-3", 5"-dimethoxyphenyl) ethyl moiety. In addition, the HMBC correlations fromH-10 to C-2 and C-3; from both H3-20 and OCH3-2 to C-2 demonstrated that C-1′ and C-3 of the two moieties were connected to the methoxy-substituted C-2 in 1. This, combined with the molecular composition and the chemical shifts of C-2 and C-7a, indicated that C-2 must connect to C-7a via a nitrogen atom to form an indolin-3- one ring. Accordingly, the planar structure of 1 is elucidated as 2- [1′-(4"-hydroxy-3", 5"-dimethoxyphenyl)ethyl]-2-methoxyindolin-π* 3-one. Although no useful information for the relative configuration was deduced from the NOE difference spectra (Fig. S22 in Supporting information), possibly due to a free rotation of C- 1′-C-2 and C-1′-C-1" bonds, the stereochemistry was proposed by comparing the experimental circular dichroism (CD) spectrum of 1 with the electronic CD (ECD) spectra predicted from the quantummechanical time-dependent density functional theory (TDDFT) calculations [36] of the four possible stereoisomers (Fig. 3). The experimental CD spectrum of 1 was in good agreement with the calculated ECD spectrum of the preassigned 1′R, 2R-isomer except that, in the long wavelength region two Cotton effect peaks at 347 and 418 nm in the experimental CD spectrum were replaced by the Cotton effect peak at 380 nm in the calculated ECD spectrum. Therefore, the structure of compound 1 was determined as (-)- (1′R, 2R)-2-[1′-(4"-hydroxy-3", 5"-dimethoxyphenyl)ethyl]-2-methoxyindolin-π* 3-one and named isatidifoliumindolinone A.

|
Download:
|
| Figure 2. Main 1H-1H COSY (thick lines) and three-bond HMBC correlations (red arrows, from 1H to 13C) of 1-4. | |
{kind=link}

|
Download:
|
| Figure 3. The overlaid experimental CD (full lines) and calculated ECD (dash lines)spectra of 1-4. | |
{kind=link}
Compound 2 was separated from 1 by HPLC using the chiral column with peak integrations of 1 and 2 in around 2:1 ratio (Fig. S7 in Supporting information). This suggested that 2 is the enantiomer of 1, which was confirmed by identity of spectroscopic features of the two compounds, but reverse specific rotations and CD curves. The 1′S, 2S-configuration of 2 was further confirmed by comparison of the experimental CD and calculated ECD spectra (Fig. 3). Therefore, the structure of 2 was determined as (+)-(1′S, 2S)-2-[1′-(4"-hydroxy-3", 5"-dimethoxyphenyl) ethyl]-2-methoxyindolin-3-one and named isatidifoliumindolinone B.
Compound 3 was obtained as a yellowish amorphous powder with [a]D 20 -70.1 (c 0.10, MeOH). Comparison of the NMR data between 3 and 1 demonstrated that OCH3-2 and H3-2′ in 3 were deshielded by DδH +0.04 and +0.18, respectively, in contrast H-4, H-5, H-6, H-10 , H-2"/6" , and OCH3-3"/5" were shielded by DδH -0.05 to -0.23. Additionally, C-2 and C-2′ in 3 were deshielded by DδC +0.6, respectively, whereas C-1′ and C-1" were shielded by DδC -0.6 and -0.7. This suggests that 3 is a diastereoisomer of 1 with the 1′R, 2S- or 1′S, 2R-configuration, which was confirmed by 2D NMR experiments (Figs. S43-S45 in Supporting information). The CD spectrum of 3 displayed Cotton effects, positive between 212 and 307 nm and negative between 307 and 387 nm, which are consistent with those in the calculated ECD spectrum of the preassigned 1′R, 2S-isomer (Fig. 3) though the Cotton effect curves in the experimental and calculated spectra could not fully match with each other. This supports that 3 has the 1′R, 2S-configuration. Therefore, the structure of compound 3 was determined as (-)- (1′R, 2S)-2-[10-(4"-hydroxy-3", 5"-dimethoxyphenyl)ethyl]-2- methoxyindolin-3-one and named isatidifoliumindolinone C.
Compound 4 was separated from 3 by chiral HPLC, with around 1:2 integration ratio of 4:3 as indicated by the corresponding peaks in the chromatogram (Fig. S8 in Supporting information). Compound 4 had the spectroscopic features fully identical to those of 3, but the reverse specific rotation and CD curve. This unequivocally indicates that 4 is the enantiomer of 3, which was also supported by comparison of the experimental CD and calculated ECD spectra (Fig. 3). Thus, the structure of 4 was determined as (+)-(1′S, 2R)-2-[10-(4"-hydroxy-3", 5"-dimethoxyphenyl) ethyl]-2-methoxyindolin-3-one and named isatidifoliumindolinone D.
Interestingly, comparison of the specific rotation data of 1-4 indicates that signs are dominated mainly by the C-10 configuration, and that the 1′R and 2S configurations have contributions of negative values to the specific rotations (1 and 3), whereas the 1′S and 2R configurations have corresponding positive contributions (2 and 4). However, comparison of the experimental CD spectra 1- 4 shows that signs of the Cotton effects are dominated by the C-2 configuration (Fig. 3). Wherein, the CD spectra of 1 and 4 possessing the 2R-configuration exhibit three negative Cotton effect peaks around 224, 259 (sh), and 295 (sh) nm with successively decreased intensities, as well as a relatively strong positive Cotton effect peak around 347 nmand a weak peak around 418 nm. In contract the CD spectra of 2 and 3 having the 2Sconfiguration show reverse Cotton effect peaks at the corresponding wavelengths 221, 264, and 294 (sh) nm. This demonstrates that chiralities of C-1′ and C-2 have significantly different contributions to the specific rotations and Cotton effects of these stereoisomers. In addition, in the experimental CD spectra of 1-4 the two Cotton effect peaks around 347 and 418 nm with the reverse signs, arising from the n-π* transition of the conjugated indolin-3-one chromophore, are replaced by the averaged Cotton effect peak at 380 nm in the calculated ECD spectra, while the three Cotton effect peaks at 224, 259 (sh), and 295 (sh) nm, due to the overlapped π-π* transitions of the substituted indoline and phenyl chromophores, are almost overlapped with the calculated Cotton effects for 1 and 2. However, the three π-π* transitions Cotton effect peaks at 221, 264, and 294 (sh) nm in the measured spectra of 3 and 4 are replaced by the averaged strong peak at 240 nm in the calculated ECD spectra. Nevertheless, the general intensities and signs of the Cotton effects in the two respective wavelength regions of the π-π* (212-307 nm) and n-π* transitions (307- 450 nm) of the experimental CD spectra are completely consistent with those of the calculated spectra. Therefore, the ECD spectroscopic calculation method is applicable for determination of the absolute configurations of 1-4. This was further supported by the ECD spectra calculations of model compounds 2-ethyl-2- methoxyindolin-3-one and 2-(4-hydroxy-3, 5-dimethoxybenzyl)- 2-methoxyindolin-3-one, which confirmed that the Cotton effects from the n-π* transition were mainly dependent upon the C-2 configuration of the indolin-3-one chromophore, and that the 4- hydroxy-3, 5-dimethoxybenzyl at C-2 of the indolin-3-one mainly influenced on the Cotton effect intensity from the n-π* transition and the Cotton effect wavelength from the π-π* transitions (Fig. S6 in Supporting information).
Because no isomerization among the stereoisomers was observed during the experiments and because artifact formation of 1-4 should produce racemic enantiomers, separation of scalemic enantiomers from the extract suggests that 1-4 are true natural products. Accordingly, compounds 1-4 represent the first examples of natural products with the novel carbon skeleton derived from coupling between the indolin-3-one and phenylethyl units though many 2-substituted 2-methoxyindolin-3-one derivatives have been chemically synthesized [37-40]. Additionally compounds 1-4 are the first natural products with all the four stereoisomers being separated as the scalemic enantiomers and stereo-chemically determined [41]. This provides support for the presence of partially stereo-controlled biosynthesis pathways in this plant, which is uncommon in nature. Based on the unique structure feature consisting of the indolin-3-one and phenylethyl moieties, the biosynthetic precursors of 1-4 are readily traced to 1H-indol-3-ol glycosides (such as isatan B, 5) and sinapic acid (6), which are abundantly co-occurring in the plants of this genus [42-47]. A plausible biosynthetic pathway for 1-4 is postulated in Scheme 1. Hydrolysis of 5 liberates tautomers 7a and 7b, which undergo nucleophilic addition with 6 to produce an intermediate 8. The intermediate can also be formed by an oxidative coupling between 5 and 6 followed by hydrolysis. Sequential and/or simultaneous enzyme-catalyzed dehydrogenation, decarboxylation, and methoxylation (or hydration then methylation) of 8 would generate 1-4. In the sequential and/or simultaneous reactions, tautomerization of the intermediates 9a/9b and 10a/10b is possibly associated with formation of the scalemic enantiomers. Alternatively (not shown in Scheme 1), compounds 1-4 would also be biosynthesized by oxidative coupling between 5 or 7a/7b with other sinapic acid-derived precursors (such as 4- hydroxy-3, 5-dimethoxyphenylethane) followed by hydration and methylation (or methoxylation).

|
Download:
|
| Scheme. 1. The plausible biosynthetic pathways of 1-4. | |
{kind=link}
In the preliminary in vitro assays carried out in this study [24-32], compounds 1-4 showed inhibitory activity against LPSinduced NO production in BV2 cells, with inhibition rates of 1.2%, 21.4%, 44.0%, and 54.0% at a concentration of 10-6 mol/L, respectively, while the positive control curcumin gave an inhibition rate of 41.2% at the same concentration. The data indicates that 3 and 4 are much more active than 1 and 2 while 4 is better than 3. This result suggests that the NO inhibition activity of the stereoisomers is highly related to the configuration at C-10 and C-2. The stereoisomers were assessed also for antiviral activity against influenza virus H1N1 PR8 and human immunodeficiency virus 1 (HIV-1), as well as against serum deprivation-π* or rotenoneinduced PC12 cell damage and several human cancer cell lines, but all were inactive at a concentration of 10-5 mol/L.
4. ConclusionFrom a decoction of da qing ye (I. in digotica leaves), the four stereoisomers of 2-[10-(4"-hydroxy-3", 5"-dimethoxyphenyl) ethyl]- 2-methoxyindolin-3-one, named isatidifoliumindolinones A-D (1-4), were isolated and stereo-chemically characterized. The stereoisomers possess the novel carbon skeleton of 2-(1-phenylethyl)- indole, which has never been found in natural and synthetic compounds. Especially the occurrence of the scalemic enantiomers in the plant extract supports their natural origin. This provides a new framework for future synthetic and biological investigations. In particular, the postulated biosynthetic pathway is a clue for biomimetic and total synthesis. In addition, it is found that the C-1′ configuration has the decisive contribution to the sign of specific rotation while the C-2 configuration determines the Cotton effect signs in the CD spectra of the stereoisomers. This provides a basic reference to assign the absolute configurations of other similar compounds. Among the stereoisomers with the druglike structures and obvious stereochemistry-activity relationship, compound 4 is most active to inhibit LPS-inducedNO production in BV2-cells, which is a promising hit deserving of further study. The present result, combined with our previous studies on the extract of “ban lan gen” [24-32], continuously demonstrates that the various chemical components, including previously unknown minor constituents with diverse structure types, as well as the scalemic and racemic stereoisomers, have contributions to pharmacological efficacy that supports the conventional applications of these herbal medicines. Moreover, separation of the bioactive scalemic and racemic enantiomers from both the leave (da qing ye) and root (ban lan gen) extracts of I. in digotica indicates that this medicinal plant produces diverse racemic and scalemic constituents, providing an area of inquiry for biogenetic mechanisms and pathways to create these natural products.
Acknowledgments Financial support from the National Natural Science Foundation of China (NNSFC; Nos. 8163"94,81373287,and 30825044) is acknowledged.| [1] | Jiangsu New Medical College, Dictionary of Traditional Chinese Medicine, Shanghai Science and Technology Publishing House, Shanghai, 1986, pp. 126-127, 1250-1252. |
| [2] | Chinese Pharmacopoeia Commission, Pharmacopoeia of People's Republic of China, Part 1, China Medical Science Press, Beijing, 2015, pp. 1032-1033, p. 21, 205. |
| [3] | Lin C.W, Tsai F.J, Tsai C.H, Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antivir. Res 68 (2005) 36–42. DOI:10.1016/j.antiviral.2005.07.002 |
| [4] | Chen L, Lin T, Zhang H.X, Immune responses to foot-and-mouth disease DNA vaccines can be enhanced by coinjection with the Isatis indigotica extract. Intervirology 48 (2005) 207–212. DOI:10.1159/000084596 |
| [5] | Liau B.C, Jong T.T, Lee M.R, LC-APCI-MS method for detection and analysis of tryptanthrin, indigo, and indirubin in Daqingye and Banlangen. J. Pharm. Biomed. Anal 43 (2007) 346–351. DOI:10.1016/j.jpba.2006.06.029 |
| [6] | Hsuan S.L, Chang S.C, Wang S.Y, The cytotoxicity to leukemia cells and antiviral effects of Isatis indigotica extracts on pseudorabies virus. J. Ethnopharm 123 (2009) 61–67. DOI:10.1016/j.jep.2009.02.028 |
| [7] | Wei X.Y, Leung C.Y, Wong C.K.C, Bisindigotin, a TCDD antagonist from the Chinese medicinal herb Isatis indigotica. J. Nat. Prod 68 (2005) 427–429. DOI:10.1021/np049662i |
| [8] | Liu J.F, Jiang Z.Y, Wang R.R, Isatisine A, a novel alkaloid with an unprecedented skeleton from leaves of Isatis indigotica. Org. Lett 9 (2007) 4127–4129. DOI:10.1021/ol701540y |
| [9] | Xu W.D, Tian Y, Guo Q.L, Secoeuphoractin, a minor diterpenoid with a new skeleton from Euphorbia micractina. Chin. Chem. Lett 25 (2014) 1531–1534. DOI:10.1016/j.cclet.2014.09.012 |
| [10] | Tian Y, Guo Q, Xu W, A Minor Diterpenoid with a new 6/5/7/3 fused-ring skeleton from Euphorbia micractina. Org. Lett 16 (2014) 3950–3953. DOI:10.1021/ol501760h |
| [11] | Song W.X, Yang Y.C, Shi J.G, Two new b-hydroxy amino acid-coupled secoiridoids from the flower buds of Lonicera japonica:isolation, structure elucidation, semisynthesis, and biological activities. Chin. Chem. Lett 25 (2014) 1215–1219. DOI:10.1016/j.cclet.2014.05.037 |
| [12] | Jiang Z.B, Jiang B.Y, Zhu C.G, Aromatic acid derivatives from the lateral roots of Aconitum carmichaelii. J. Asian Nat. Prod. Res 16 (2014) 891–900. DOI:10.1080/10286020.2014.939585 |
| [13] | Jiang Z.B, Song W.X, Shi J.G, Two new 1-(60-O-acyl-b-D-glucopyranosyl)pyridinium-3-carboxylates from the flower buds of Lonicera japonica. Chin. Chem. Lett 26 (2015) 69–72. DOI:10.1016/j.cclet.2014.10.011 |
| [14] | Yu Y, Jiang Z, Song W, Glucosylated caffeoylquinic acid derivatives from the flower buds of Lonicera japonica. Acta Pharm. Sin. B 5 (2015) 210–214. DOI:10.1016/j.apsb.2015.01.012 |
| [15] | Guo Q, Wang Y, Lin S, 4-Hydroxybenzyl-substituted amino acid derivatives from Gastrodia elata. Acta Pharm. Sin. B 5 (2015) 350–357. DOI:10.1016/j.apsb.2015.02.002 |
| [16] | Guo Q.L, Wang Y.N, Zhu C.G, 4-Hydroxybenzyl-substituted glutathione derivatives from Gastrodia elata. J. Asian Nat. Prod. Res 17 (2015) 439–454. DOI:10.1080/10286020.2015.1040000 |
| [17] | Jiang Y, Liu Y, Guo Q, Acetylenes and fatty acids from Codonopsis pilosula. Acta Pharm. Sin. B 5 (2015) 215–222. DOI:10.1016/j.apsb.2015.03.005 |
| [18] | Jiang Y.P, Liu Y.F, Guo Q.L, C14-Polyacetylene glucosides from Codonopsis pilosula. J. Asian Nat. Prod. Res 17 (2015) 601–614. DOI:10.1080/10286020.2015.1041932 |
| [19] | Jiang Z.B, ${referAuthorVo.mingEn} X.H.Meng, Jiang B.Y, Two 2-(quinonylcarboxamino)benzoates from the lateral roots of Aconitum carmichaelii. Chin. Chem. Lett 26 (2015) 653–656. DOI:10.1016/j.cclet.2015.04.011 |
| [20] | Jiang Y.P, Guo Q.L, Liu Y.F, Codonopiloneolignanin A, a polycyclic neolignan with a new carbon skeleton from the roots of Codonopsis pilosula. Chin. Chem. Lett 27 (2016) 55–58. DOI:10.1016/j.cclet.2015.11.009 |
| [21] | Jiang Y.P, Liu Y.F, Guo Q.L, Sesquiterpene glycosides from the roots of Codonopsis pilosula. Acta Pharm. Sin. B 6 (2016) 46–54. DOI:10.1016/j.apsb.2015.09.007 |
| [22] | Meng X.H, Jiang Z.B, Zhu C.G, Napelline-type C20-diterpenoid alkaloid iminiums from an aqueous extract of "fu zi":solvent-/base-/acid-dependent transformation and equilibration between alcohol iminium and aza acetal forms. Chin. Chem. Lett 27 (2016) 993–1003. DOI:10.1016/j.cclet.2016.05.013 |
| [23] | Q.L. Guo, S. Lin, Y.N. Wang, et al., Gastrolatathioneine, an unusual ergothioneine derivative from an aqueous extract of "tian ma":a natural product co-produced by plant and symbiotic fungus, Chin. Chem. Lett. Chin. Chem. Lett. (2016), http://dx.doi.org/10.1016/j.cclet.2016.06.040. |
| [24] | Chen M, Gan L, Lin S, Alkaloids from the root of Isatis indigotica. J. Nat. Prod 75 (2012) 1167–1176. DOI:10.1021/np3002833 |
| [25] | Chen M, Lin S, Li L, Enantiomers of an indole alkaloid containing unusual dihydrothiopyran and 1, 2,4-thiadiazole rings from the root of Isatis indigotica. Org. Lett 14 (2012) 5668–5671. DOI:10.1021/ol302660t |
| [26] | Wang X, Chen M, Wang F, Chemical constituents from root of Isatis indigotica. Chin. J. Chin. Mater. Med 38 (2013) 1172–1182. |
| [27] | Liu Y.F, Chen M.H, Wang X.L, Antiviral enantiomers of a bisindole alkaloid with a new carbon skeleton from the roots of Isatis indigotica. Chin. Chem. Lett 26 (2015) 931–936. DOI:10.1016/j.cclet.2015.05.052 |
| [28] | Liu Y.F, Chen M.H, Guo Q.L, Antiviral glycosidic bisindole alkaloids from the roots of Isatis indigotica. J. Asian Nat. Prod. Res 17 (2015) 689–704. DOI:10.1080/10286020.2015.1055729 |
| [29] | Liu Y.F, Chen M.H, Lin S, Indole alkaloid glucosides from the roots of Isatis indigotica. J. Asian Nat. Prod. Res 18 (2016) 1–12. DOI:10.1080/10286020.2015.1117452 |
| [30] | Liu Y.F, Wang X.L, Chen M.H, Three pairs of alkaloid enantiomers from the root of Isatis indigotica. Acta Pharm. Sin. B 6 (2016) 141–147. DOI:10.1016/j.apsb.2016.01.003 |
| [31] | Chen M.H, Lin S, Wang Y.N, Antiviral stereoisomers of 3,5-bis(2-hydroxybut-3-en-1-yl)-1,2,4-thiadiazole from the roots Isatis indigotica. Chin. Chem. Lett 27 (2016) 643–648. DOI:10.1016/j.cclet.2016.01.042 |
| [32] | Y.F. Liu, M.H. Chen, Q.L. Guo, et al., Antiviral aromatic metabolites from "ban lan gen", Acta Pharm. Sin. B (2016) (in press). |
| [33] | MOE 2009.10, Chemical Computing Group Inc., Montreal, Canada, 2009. |
| [34] | Gaussian 09, Revision C.01, Gaussian Inc., Wallingford, CT, 2009; A full list of authors can be found in the Supporting Information |
| [35] | Wu X, Liu Y, Sheng W, Chemical constituents of Isatis indigotica. Planta Med 63 (1997) 55–57. DOI:10.1055/s-2006-957604 |
| [36] | Li X.C, Ferreira D, Ding Y, Determination of absolute configuration of natural products:theoretical calculation of electronic circular dichroism as a tool. Curr. Org. Chem 14 (2010) 1678–1697. DOI:10.2174/138527210792927717 |
| [37] | Chien C.S, Suzuki T, Kawasaki T, Oxidation of l-acylindoles with oxodiperoxomolybdenum (VI), MoO5 HMPA. Preparation of 2,3-dihydroxyindoline and indoxyl derivatives. Chem. Pharm. Bull 32 (1984) 3945–3951. DOI:10.1248/cpb.32.3945 |
| [38] | Kawasaki T, Nonaka Y, Kobayashi M, Convenient procedure for the synthesis of 2-monoalkylated indol-3-ones. J. Chem. Soc. Perkin Trans 1 (1991) 2445–2448. |
| [39] | Higuchi K, Masuda K, Koseki T, Asymmetric alkylation of 2-monosubstituted indolin-3one. Heterocycles 73 (2007) 641–650. DOI:10.3987/COM-07-S(U)42 |
| [40] | Lerch S, Unkel L.N, Brasholz M, Tandem organocatalysis and photocatalysis:an anthraquinone-catalyzed indole-C3-alkylation/photooxidation/1,2-shift sequence. Angew. Chem. Int. Ed 53 (2014) 6558–6562. DOI:10.1002/anie.201402920 |
| [41] | Finefield J.M, Sherman D.H, Kreitman M, Enantiomeric natural products:occurrence and biogenesis. Angew. Chem. Int. Ed 51 (2012) 4802–4836. DOI:10.1002/anie.v51.20 |
| [42] | Epstein E, Nabors M.W, Stowe B.B, Origin of indigo of woad. Nature 216 (1967) 547–549. DOI:10.1038/216547a0 |
| [43] | Maugarda T, Enauda E, Choisy P, Identification of an indigo precursor from leaves of Isatis tinctoria (Woad). Phytochemistry 58 (2001) 897–904. DOI:10.1016/S0031-9422(01)00335-1 |
| [44] | Oberthür C, Schneider B, Graf H, The elusive indigo precursors in woad (Isatis tinctoria L.)-identification of the major indigo precursor, isatan A, and a structure revision of isatan B. Chem. Biodiv 1 (2004) 174–182. DOI:10.1002/(ISSN)1612-1880 |
| [45] | Oberthür C, Graf H, Hamburger M, The content of indigo precursors in Isatis tinctoria leaves-a comparative study of selected accessions and post-harvest treatments. Phytochemistry 65 (2004) 3261–3268. DOI:10.1016/j.phytochem.2004.10.014 |
| [46] | Tozzi S, Lercari B, Angelini L.G, Light quality influences indigo precursors production and seed germination in Isatis tinctoria L. and Isatis indigotica Fort. Photochem. Photobiol 81 (2005) 914–919. DOI:10.1562/2004-08-03-RA-258R1.1 |
| [47] | Mohn T, Plitzko I, Hamburger M, A comprehensive metabolite profiling of Isatis tinctoria leaf extracts. Phytochemistry 70 (2009) 924–934. DOI:10.1016/j.phytochem.2009.04.019 |