 2016, Vol. 27
2016, Vol. 27 


 , Ming Chengb
, Ming Chengb
b Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China ;
c University of the Chinese Academy of Sciences, Beijing 100049, China ;
d School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, China
Organophosphates have attracted increasingly synthetic pursuit of chemists because of their widely applications in many major physiological processes [1], drug discovery [2], organic synthesis [3] and agrochemicals [4]. Particularly, α-hydroxy-ketone phosphates can be used as sugar analogues [5] and important intermediates for the construction of phospholipid and oligonucleotide through the selective hydrolytic removal of the ketoxide motif [6]. As such, the development of general and efficient methods to access α-hydroxyketone phosphates is of great interest. Traditionally, α-hydroxyketone phosphates are synthesized by the α-phosphoryloxylation of ketones with the [hydroxy(phosphoryloxy)iodo]arenes [7], the reaction of 2, 2, 2-trialkoxy-1, 3, 2-dioxaphospholen with hydrogen chloride [5], and the oxyphosphorylation of silyl enol ethers with phosphoric acid and p-(difluoroiodo) toluene [8]. An alternative method for the construction of α-hydroxyketone phosphates has also been developed via the addition of terminal alkynes with unstable hypervalent iodine compound intermediate, which was preformed from the reaction of phosphonic acid with iodosobenzene (Scheme 1, Eq. (1)) [9]. However, all these methods suffer from limitations such as unreadily available starting materials, tedious work-up procedures, relatively harsh reaction conditions, toxic chemical wastes, the poor substrate scope, or low yields. Therefore, it is still highly desirable to develop a simple, convenient and efficient approach to α-hydroxyketone phosphates.

|
Download:
|
| Scheme. 1. Methods for the synthesis of α-hydroxyketone phosphates. | |
In 2012, Yan [10] and co-workers reported iodobenzene/mchloroperbenzoic acid (m-CPBA) mediated the α-phosphoryloxylation of ketones with (RO)2PO2H (Scheme 1, Eq. (2)). Very recently, Wang et al. [11] reported a new method for the construction of α-hydroxyketone phosphates through I2O5/DBU mediated direct α-phosphoryloxylation of ketones with Hphosphonates (Scheme 1, Eq. (3)). Nevertheless, stoichiometric amount of potentially dangerous peroxide oxidant or base are still required in the two well developed reactions. Here, we wish to report a simple, convenient and highly efficient PhI(OAc)2 mediated procedure for the construction of α-hydroxyketone phosphates from alkynes and H-phosphine oxides in the presence of water under mild conditions (Scheme 1, Eq. (4)). The present protocol provides an alternative and highly attractive route to various α-hydroxyketone phosphates from the commercially available starting materials, and especially it avoids the use of unstable reagents, and stoichiometric amounts of bases, toxic or potentially dangerous oxidants.
2. ExperimentalAll chemicals and solvents were purchased from Aldrich, J & K and Alfa Aesar Chemical Company as reagent grade and used without further purification unless otherwise stated. 1H NMR and 13C NMR spectra were collected in CDCl3 on a Bruker Avance 400 spectrometer with TMS as internal standard at room temperature, 31P NMR spectra were recorded at 162 MHz, and chemical shifts (δ) reported relative to external 85% phosphoric acid (δ=0.0 ppm), the chemical shifts (δ) were expressed in parts per million (ppm) and J values were given in hertz (Hz). HRMS were performed on a Brucker Daltonics Bio-TOF-Q mass spectrometer by the ESI method and LC-MS were obtained on a on a Waters Xevo TQ (Waters, Manchester, UK) equipped with an ESI source. The products were purified by flash column chromatography on silica gel (200-300 mesh).
2.1. General procedure for the synthesis of α-hydroxyketone phosphates 3To a solution of diarylphosphine oxides 2 (0.5 mmol) in acetonitrile (3.0 mL) were added PhI(OAc)2 (1.25 mmol), H2O (2.0 mmol) and alkynes 1 (0.75 mmol). The reaction mixture was then stirred for 24 h at 60 ℃ in air. After the reaction, the solvents were removed under vacuum. The residue was purified by flash chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (1:1) as eluent to give the desired product 3.
2.2. The procedure of gram-scale reaction for the synthesis of 3aaTo a solution of diphenylphosphine oxide 2a (2.02 g, 10.0 mmol) in acetonitrile (60.0 mL) were added PhI(OAc)2 (8.05 g, 25.0 mmol), H2O (720.0 μL, 40.0 mmol) and phenylacetylene 1a (1.53 g, 15.0 mmol). The reaction mixture was then stirred for 24 h at 60 ℃ in air. After the reaction, the solvents were removed under vacuum. Water (60.0 mL) was added to the reaction mixture, and then the mixture was extracted with EtOAc. The combined organic phase was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (1:1) as eluent to give the desired product 3aa (2.99 g, 89%).
2.3. The reaction of diphenylphosphine oxide 2a with PhI(OAc)2To a solution of diphenylphosphine oxide 2a (1.0 mmol) in acetonitrile (6.0 mL) were added PhI(OAc)2 (1.0 mmol) and H2O (4.0 mmol). The reaction mixture was then stirred for 24 h at 60 ℃ in air. After the reaction, the solvents were removed under vacuum. The residue was purified by flash chromatography on silica gel using a mixture of dichloromethane and methanol (5:1) with addition of AcOH (1‰) as eluent to give the desired product 6a (209.0 mg, 96%).
2.4. The reaction of phenylacetylene 1a with diphenylphosphinic acid 6aTo a solution of diphenylphosphinic acid 6a (0.5 mmol) in acetonitrile (3.0 mL) were added PhI(OAc)2 (1.25 mmol), H2O (2.0 mmol) and phenylacetylene 1a (0.75 mmol). The reaction mixture was then stirred for 24 h at 60 ℃ in air. After the reaction, the solvents were removed under vacuum. The residue was purified by flash chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (1:1) as eluent to give the desired product 3aa (153.0 mg, 91%).
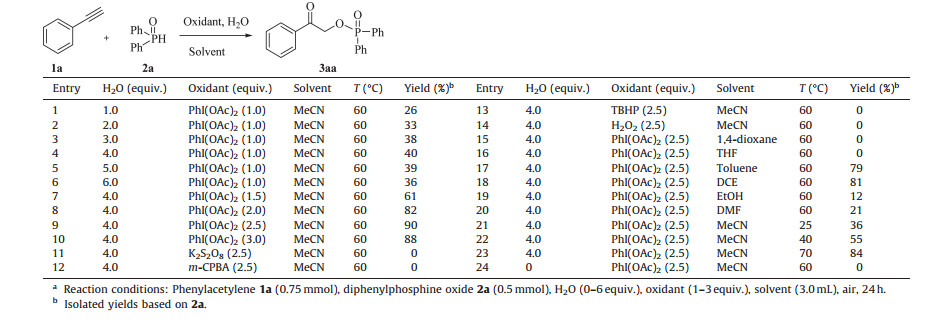
3. Results and discussionAt the outset of our investigation, the reaction of phenylacetylene 1a and diphenylphosphine oxide 2a was chosen as the model reaction to optimize the reaction conditions. Gratifyingly, the desired product 3aa was obtained in 26% yield when the model reaction was performed in the presence of PhI(OAc)2 (1.0 equiv.)/ H2O (1.0 equiv.) at 60 ℃ in air for 24 h (Table 1, entry 1). It was found that the reaction gave a better yield 40% when the loading of H2O was increased to 4.0 equiv. (Table 1, entry 4). Further optimization suggested that the reaction efficiency was obviously improved with the increasing of PhI(OAc)2 loading, the best yield was obtained when 2.5 equivalent of PhI(OAc)2 was used (Table 1, entry 9). The screening of other oxidants, such as K2S2O8, m-CPBA, TBHP and H2O2 could not improve the reaction efficiency (Table 1, entries 11-14). Subsequent investigation on the effect of solvents showed that the reaction performed in MeCN was found to be superior for the formation of 3aa (Table 1, entries 15-20). In addition, we found that the reaction temperature also played an important role in this transformation (Table 1, entries 9, 21-23). The desired product 3aa was isolated in only 36% yield when the model reaction was carried out at room temperature (Table 1, entry 21), and the best yield was obtained when the reaction was conducted at 60 ℃. It should be noted that none of desired product 3aa was detected when the reaction performed in the absence of H2O, suggesting that H2O could play the key role in the synthesis of α-hydroxyketone phosphates (Table 1, entry 24).
|
|
Table 1 Optimization of reaction conditions.a |
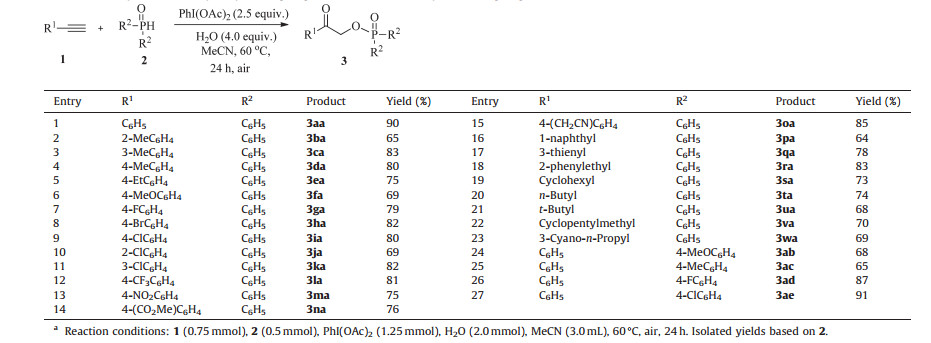
With the optimal conditions in hand, we next examined the substrate scope of this transformation. In general, both electronrich and electron-deficient aromatic alkynes were all suitable for this reaction to afford α-hydroxyketone phosphates in good to high yields under the standard conditions (Table 2, 3aa-3oa). The substituent groups on the ortho-position of aromatic ring led to a negative effect on the reaction efficiency, which might be caused by the steric hindrance (Table 2, 3ba and 3ja). Also, functional groups such as halogen, trifluoromethyl, nitro, cyano, and carboxylic ester were all well tolerated, whose corresponding products can be applied in further modifications (Table 2, 3ga-3oa). It was observed that 1-ethynylnaphthalene and heteroaromatic alkenes such as 3-ethynylthiophene were also tolerated to afford the product 3pa and 3qa in 64% and 78% yields, respectively. Notably, when aliphatic alkynes were used as the substrates, the corresponding products were also obtained in good yields (Table 2, 3ra-3wa). With respect to the H-phosphine oxides, in addition to diphenylphosphine oxide 2a, other diarylphosphine oxides bearing both electron-donating and electron-withdrawing groups were all suitable substrates, leading to the corresponding products in good to excellent yields (Table 2, 3ab-3ae).
|
|
Table 2 Results for the synthesis of α-hydroxyketone phosphates from terminal alkynes and H-phosphine oxides.a |
{kind=link}
Furthermore, the synthetic applicability of this method was investigated on a gram scale by using the model reaction between 1a and 2a. As shown in Scheme 2, the reaction could afford 2.99 g of 3aa in 89% yield without any significant loss of its efficiency. Thus, this reaction could serve as a practical and efficient protocol to synthesize α-hydroxyketone phosphates.

|
Download:
|
| Scheme. 2. Gram scale reaction. | |
{kind=link}
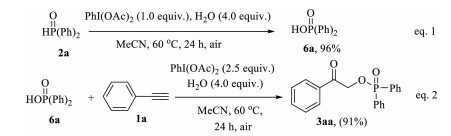
In order to gain some insights into this reaction mechanism, two control experiments were conducted (Scheme 3). When diphenylphosphine oxide 2a was independently treated with PhI(OAc)2 (1.0 equiv.)/H2O (4.0 equiv.) in the absence of phenylacetylene, the diphenylphosphinic acid 6a was obtained in 96% yield (Scheme 3, Eq. (1)). Furthermore, the desired product 3aa could be isolated in 91% yield when the reaction of diphenylphosphinic acid 6a and phenylacetylene 1a was performed under standard conditions (Scheme 3, Eq. (2)). The above results indicated that diphenylphosphinic acid 6a might be a key intermediate in the present reaction system.

|
Download:
|
| Scheme. 3. Control experiments. | |
{kind=link}
On the basis of the above results and previous reports [9, 12-14], a tentative reaction pathway was proposed in Scheme 4. Initially, diphenylphosphine oxide 2a was oxidized by phenyliodine diacetate to give diphenylphosphinic acid 6a through the consecutive transformation of the intermediate 4a and 5a [12]. Subsequently, the resulting diphenylphosphinic acid 6a reacted with phenyliodine diacetate to form the unstable hypervalent iodine intermediate 7a [13]. Next, the selective addition of hypervalent iodine reagent 7a to phenylacetylene 1a would lead to the formation of 8a, which underwent isomerization to give hypervalent iodine intermediate 9a [9, 14]. Finally, the reductive elimination of hypervalent iodine intermediate 9a produced the desired product 3aa with the release of indobenzene [9]. To our delight, the proposed intermediate 4a, 5a, 6a, and 7a were all detected by LC-MS analysis when the model reaction was performed at 1.5 h (Fig. 1 and Supporting information for detailed description of LC-MS analysis experiment).

|
Download:
|
| Scheme. 4. Possible reaction pathway. | |
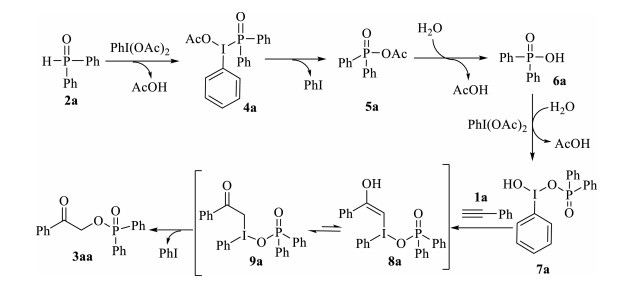{kind=link}

|
Download:
|
| Figure 1. LC-MS spectrum of 4a, 5a, 6a, 7a and 3aa. | |
{kind=link}
To gain further understanding of the detailed reaction process, the model reaction was monitored by 31P NMR spectroscopy (Fig. 2). In a round-bottom flask, a mixture of diphenylphosphine oxide 2a, PhI(OAc)2, phenylacetylene 1a and H2O was added into CD3CN. The sample was immediately drawn off and tested by 31P NMR spectrum. The spectrum showed a signal at 19.06 ppm, which was assigned as the starting material diphenylphosphine oxide 2a [15]. After being heated at 60 ℃ for 10 min, the signal of 2a disappeared, while two new signals at 29.02 and 25.20 ppm appeared, which were assigned to be diphenylphosphinic acid 6a [16] and intermediate 7a [9, 13], respectively. Then, after being heated for 20 min, the signal of diphenylphosphinic acid 6a slumped while the signal of intermediate 7a increased dramatically. Moreover, a signal at 34.09 ppm was simultaneously observed, corresponding to signals of the final product 3aa [9]. With the progress of the reaction, the 31P NMR signal of intermediate 7a disappeared gradually and the signal of 3aa increased. Thus, these data support the mechanistic hypothesis described in Scheme 4.

|
Download:
|
| Figure 2. 31P NMR spectral changes during the reaction process. | |
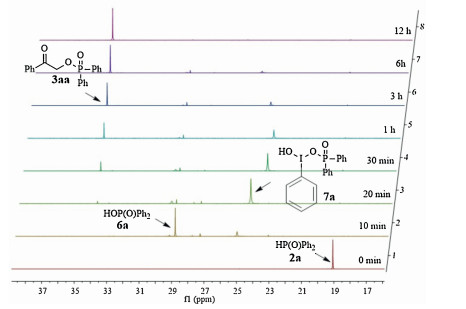{kind=link}
4. Conclusion
In conclusion, a new and simple method has been developed for the one-pot construction of α-hydroxyketone phosphates from terminal alkynes and H-phosphine oxides in the presence of PhI(OAc)2/H2O. This protocol provides a convenient and efficient approach to various α-hydroxyketone phosphates in good to high yields from commercially available starting materials with high regioselectivity and excellent functional group tolerance. Such an attractive synthesis methodology for α-hydroxyketone phosphates would find the potential applications in the fields of synthetic and pharmaceutical chemistry. The detailed scope, mechanism, and synthetic application of this reaction are currently underway in our laboratory.
AcknowledgmentsWe gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos. 21172213, 21302109, 21402184, and 21572217).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.05.029.
| [1] | M.W. Bowler, M.J. Cliff, J.P. Waltho, G.M. Blackburn, Why did nature select phosphate for its dominant roles in biology. N. J. Chem. 34 (2010) 784–794. DOI:10.1039/b9nj00718k |
| [2] |
(a) C. Schultz, Prodrugs of biologically active phosphate esters, Bioorg. Med. Chem. 11(2003) 885-898; (b) C.Ducho, U.Gorbig, S. Jessel, etal., Bis-cycloSal-d4T-monophosphates: drugs that deliver two molecules of bioactive nucleotides, J. Med. Chem. 50(2007) 1335-1346; (c) S.J. Hecker, M.D. Erion, Prodrugs of phosphates and phosphonates, J. Med. Chem. 51(2008) 2328-2345. |
| [3] |
(a) S. Protti, M. Fagnoni, Phosphate esters as "tunable" reagents in organic synthesis, Chem. Commun. 31(2008) 3611-3621; (b) A. Parra, S. Reboredo, A.M. Martin Castro, J. Aleman, Metallic organophosphates as catalysts in asymmetric synthesis: a return journey, Org. Biomol. Chem. 10(2012) 5001-5020. |
| [4] |
(a) R. Neumann, H.H. Peter, Insecticidal organophosphates: nature made them first, Experientia 43(1987) 1235-1237; (b) H.W. He, The use of OP and development of new organophosphorus agrochemicals in China, Phosphorus Sulfur Silicon Relat. Elem. 183(2008) 266-279; (c) J.Wink, F.R.Schmidt, G.Seibert, W.Aretz, Cyclipostins: novel hormone-sensitive lipase inhibitors from Streptomyces sp. DSM 13381, I. Taxonomic studies of the producer microorganism and fermentation results, J. Antibiot. 55(2002) 472-479; (d) L. Vìrtesy, B. Beck, M. Bronstrup, et al., Cyclipostins, novel hormone-sensitive lipase inhibitors from Streptomyces sp. DSM 13381, Ⅱ. Isolation, structure elucidation and biological properties, J. Antibiot. 55(2002) 480-494. |
| [5] | F. Ramirez, J. Bauer, C.D. Telefus, Introduction of the amide function into 1, 3, 2-dioxaphospholenes with pentavalent phosphorus. J. Am. Chem. Soc. 92 (1970) 6935–6942. DOI:10.1021/ja00726a035 |
| [6] |
(a) F. Ramirez, B. Hansen, N.B. Desai, Kinetics and mechanisms of the rapid alkaline hydrolysis of dimethylphosphoacetoin, J. Am. Chem. Soc. 84(1962) 4588; (b) H. Witzel, A. Botta, K. Dimroth, Mechanismus der alkalischen hydrolyse von dialkyl-[2-oxo-alkyl]-phosphaten, Chem. Ber. 98(1965) 1465-1469; (c) R. Kluger, S.D. Taylor, Mechanisms of carbonyl participation in phosphate ester hydrolysis and their relationship to mechanisms for the carboxylation of biotin, J. Am. Chem. Soc. 113(1991) 996-1001; (d) F. Ramirez, J.F. Marecek, Synthesis of phosphodiesters: the cyclic enediol phosphoryl (CEP) method, Synthesis (1985) 449-488. |
| [7] |
(a) F. Ramirez, N.B. Desai, Crystalline 1:1 adducts from the reaction of tertiary phosphate esters with ortho-quinones and with alpha-diketones. New routes to quinol-monophosphates and to ketol-monophosphates, J. Am. Chem. Soc. 82(1960) 2652-2653; (b) F. Ramirez, S.L. Glaser, A.J. Bigler, J.F. Pilot, Synthesis of sugar-like phosphates by the oxyphosphorane condensation. Reaction of glyoxal with trialkyl phosphites and preparation of phosphate esters of glycolaldehyde, a-hydroxy b-keto aldehydes, and hydroxy malon-aldehyde chloride, J. Am. Chem. Soc. 91(1969) 496-500; (c) F. Ramirez, S.B. Bhatia, A.J. Bigler, C.P. Smith, New syntheses of β-keto-α-hydroxy acid chlorides, of α-hydroxy β-diketones, and of their phosphate esters, J. Org. Chem. 33(1968) 1192-1196; (d) G.F. Koser, J.S. Lodaya, D.G. Ray, P.B. Kokil, Direct α-phosphoryloxylation of ketones and phosphoryloxylactonization of pentenoic acids with[hydroxy((bis(pheny1oxy)phosphory1)oxy)-ido]benzene, J. Am. Chem. Soc. 110(1988) 2987-2988; (e) T. Nabana, H. Togo, Reactivities of novel[hydroxy (tosyloxy)iodo]-arenes and[hydroxy(phosphoryloxy)iodo]arenes for a-tosyloxy lation and a-phosphoryloxylation of ketones, J. Org. Chem. 67(2002) 4362-4365. |
| [8] | G.F. Koser, K.C. Chen, Y.L. Huang, C.A. Summers, Oxyphosphorylation of carbon with phosphoric acid and p-(dif1uoroiodo)toluene: synthesis of tris-ketol phosphates and their conversion into lithium bis-ketol phosphates. J. Chem. Soc. Perkin Trans. 1 (1994) 1375–1376. |
| [9] | R.M. Morisrty, C. Condeiu, A. Tao, O. Prskash, New organohyper-valent iodine reagents for α-methylphosphonylations and α-diphenyl-and a-dimethylphosphinylations. Tetrahedron Lett. 38 (1997) 2401–2404. DOI:10.1016/S0040-4039(97)00388-2 |
| [10] | Y. Pu, L.M. Gao, H.J. Liu, J. Yan, An effective catalytic α-phosphoryloxylation of ketones with iodobenzene. Synthesis 44 (2011) 99–103. |
| [11] | C.L. Liu, W. Wei, D.S. Yang, I2O5/DBU mediated direct α-phosphoryloxylation of ketones with H-phosphonates leading to α-hydroxyketone phosphates. Tetrahedron 71 (2015) 6901–6906. DOI:10.1016/j.tet.2015.07.017 |
| [12] | A. Hubacz, S. Makowiec, Direct conversion of secondary phosphine oxides and Hphosphinates with [di(acyloxy)iodo]benzenes to phosphinic and phosphonic amides. Heteroatom Chem. 20 (2009) 81–86. DOI:10.1002/hc.v20:2 |
| [13] | S.A. Moteki, A. Usui, T. Zhang, C.R. Solorio Alvarado, K. Maruoka, Site-selective oxidation of unactivated Csp3-H bonds with hypervalent iodine (Ⅲ) reagents. Angew. Chem. Int. Ed. 125 (2013) 8819–8822. DOI:10.1002/ange.201304359 |
| [14] | S. Khamarui, R. Maiti, R.R. Mondal, D.K. Maiti, Reactant cum solvent water: generation of transient λ3-hypervalent iodine, its reactivity, mechanism and broad application. RSC Adv. 5 (2015) 106633–106643. DOI:10.1039/C5RA21932A |
| [15] |
(a) A. Christiansen, C. Li, M. Garland, et al., On the tautomerism of secondary phosphane oxides, Eur. J. Org. Chem. 14(2010) 2733-2741; (b) A.A. Bobrikova, M.P. Koroteev, A.M. Koroteev, Y.V. Nelyubina, E.E. Nifant'ev, Phosphorylation of N-glycosides derived from para-substituted aromatic amines, Russ. Chem. Bull 57(2008) 2021-2027. |
| [16] | O.I. Artyushin, E.V. Sharova, P.V. Petrovskii, I.L. Odinets, N-Alkylation of amidothiophosphoryl compounds under phase-transfer catalysis conditions. Synthesis and properties of 1, 3, 2-thiazaphos-phacyclanes. Russ. Chem. Bull. 58 (2009) 216–222. DOI:10.1007/s11172-009-0032-4 |